

In the currently valid version of the Medical Device Regulation (MDR), there is considerable room for interpretation when it comes to classifying software medical devices. In particular, the distinction between risk classes I and IIa is vaguely worded. Manufacturers, authorities, and notified bodies have different interpretations of this.
On December 16, 2025, the EU Commission published a proposed amendment to the Medical Device Regulation (MDR). Among other things, this draft amendment includes a change to the notorious Rule 11 on the classification of software medical devices.
The big question for manufacturers of digital health applications (DiGA) and medical software: Which software products fall under the new draft in Class I, and which in Class IIa?
According to the EU Commission, one of the objectives of this proposed amendment to the MDR is to promote innovation. It therefore stands to reason that the new draft will now clearly stipulate that low-risk products also fall into risk class I. As you will read below, this is unfortunately not the case.
We have examined the draft and all relevant documents in detail. In this article, we provide answers to questions about the currently planned classification of software medical devices.
Table of Contents
- 1. Current Status: Classification of Software Medical Devices
- 2. Planned Draft: Amendment to Rule 11 for Software Medical Devices
- 3. Interpretations of the MDR Amendments on Risk Classification
- 4. What else falls into Risk Class I with the Draft Amendments to the MDR?
- 5. Is this proposed change final and will it be implemented as such?
- 6. Timeline: When will the MDR changes and the new Rule 11 take effect?
- 7. Further Interpretations
- 8. Conclusion
1. Current Status: Classification of Software Medical Devices
Until the planned changes are incorporated into the MDR in some form, the current provisions will of course continue to apply for the time being. In this context, we have written three detailed guides to help software medical device manufacturers classify their products into an MDR risk class:
- Classification of software medical devices: MDR Guideline
- Class I software according to MDR – Is that still possible?
- MDR Risk Class I vs. IIa: Differences for software medical devices
2. Planned Draft: Amendment to Rule 11 for Software Medical Devices
Rule 11 is arguably the most important part of the MDR in terms of the classification of software medical devices. This rule is now set to undergo fundamental changes.
The current draft of this amendment can be found here: Annex to the Proposal for a regulation to simplify rules on medical and in vitro diagnostic devices
The new wording of Rule 11 would then read as follows:
6.3 Rule 11
Software which is intended to generate an output that confers a clinical benefit and is used for diagnosis, treatment, prevention, monitoring, prediction, prognosis, compensation or alleviation of a disease or condition is classified as class I, unless the output is intended for a disease or condition:
- in a critical situation with a risk of causing death or an irreversible deterioration of a person’s state of health, in which case it is classified as class III;
- in a serious situation with a risk of causing a serious deterioration of a person’s state of health or a surgical intervention, or to drive clinical management in a critical situation in which cases it is classified as class IIb;
- in a non-serious situation, or to drive clinical management in a serious situation or to inform clinical management in a critical or serious situation in which cases it is classified as class IIa.’;
According to the proposed rule, all software will initially be classified as Class I, unless criteria for a higher classification apply. This is a reversal of the previous logic and, from a manufacturer’s perspective, sounds welcome at first glance. It almost seems as if Class I is the rule rather than the exception for software medical devices. However, upon closer inspection, this impression is not necessarily confirmed.
3. Interpretations of the MDR Amendments on Risk Classification
Our team of experts came up with two possible interpretations when analyzing the proposed Rule 11.
In addition to the plain wording, we also use the following documents to interpret the rule:
- MDCG 2019-11 Rev.1: The quasi-official guidance document on the classification and qualification of software medical devices under the MDR. Of particular relevance here is the table in Annex III, which refers to the IMDRF risk classification.
- IMDRF – Software as a Medical Device: Possible Framework for Risk Categorization and Corresponding Considerations: International framework for risk-based classification of SaMD. It evaluates software based on the clinical significance of the information generated and the severity of the medical situation and forms the conceptual basis for software classification in MDCG 2019-11.
These documents help to understand key terms (e.g., “critical situation,” “drive clinical management”) in the proposed MDR amendment and include the tables used below.
3.1 Interpretation 1: Strict Interpretation
Strictly speaking, the rule can be interpreted to mean that any software that
- … is used in a “critical situation,” falls into Class III.
- … is used in a “serious situation,” falls under Class IIb.
- … is used in a “non-serious situation,” falls under Class IIa.
Any exceptions (e.g., “to drive clinical management in a critical situation […] is classified as class IIb”) are not taken into account in this interpretation. According to the MDR, the strictest applicable (sub)rule always applies with regard to classification. Accordingly, the new rule could be interpreted to mean that the general rule for classification in the higher risk class always applies.
According to this interpretation, the following classification of risk classes would be arrived at:
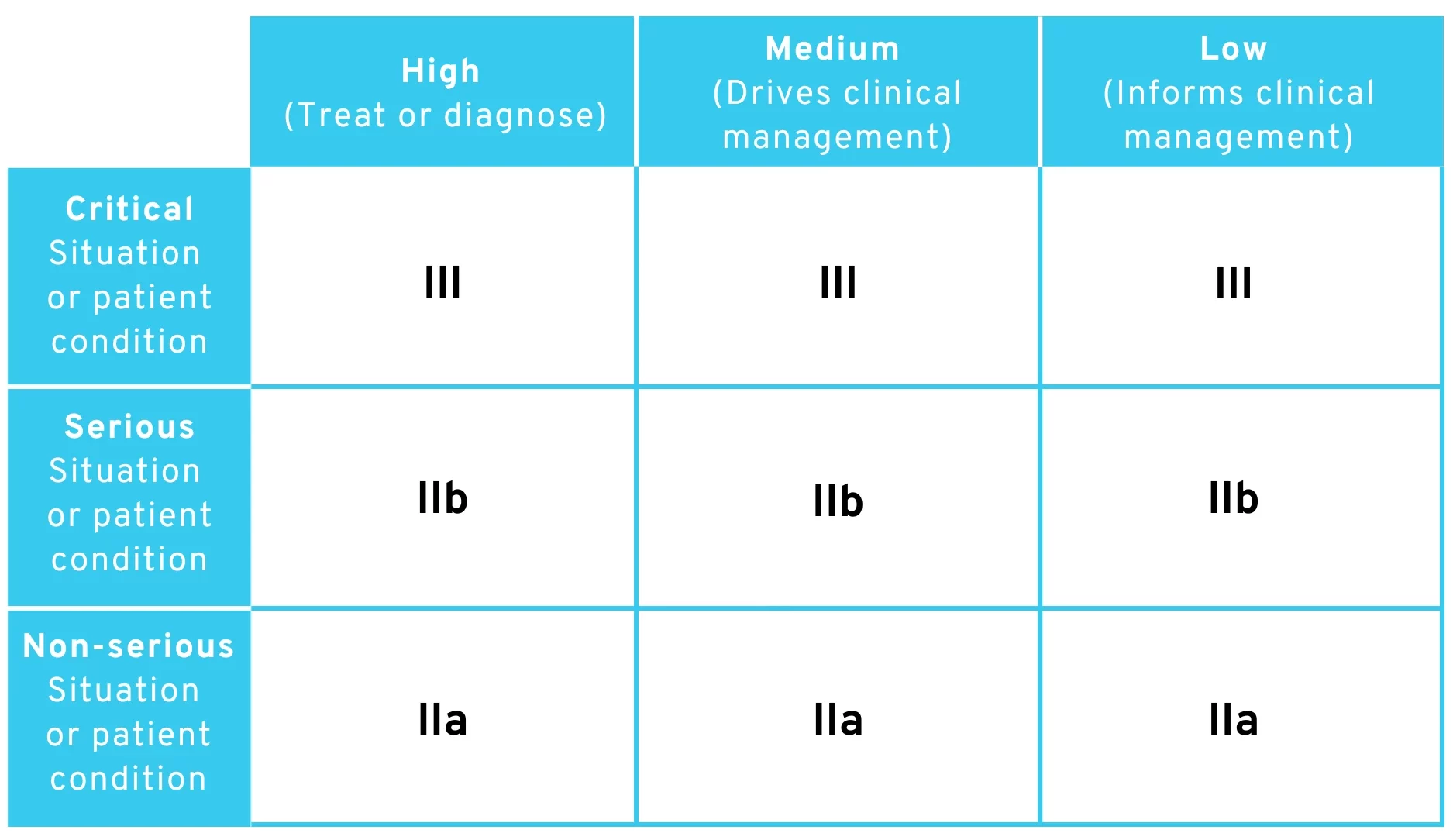
Proposed amendment to MDR Rule 11 – Table for classification of risk classes with strict interpretation
As you can see, risk class I is completely missing here. There is no obvious argument for still assigning software to class I in this case.
However, we consider this interpretation to be highly unlikely, as the wording of Rule 11 could be much simpler in this case and the EU Commission certainly had something in mind when it came to the exceptions and additions. From a legal perspective, it is also questionable whether the sub-items of Rule 11 can be described as “sub-rules” within the meaning of MDR Implementing Rule 3.5 (according to which the strictest sub-rule on classification must always be applied).
3.2 Interpretation 2: Literal Interpretation
At first glance, the rule could be interpreted in the same way as above. However, if we also take into account the explicitly stated exceptions (e.g., “to drive clinical management in a critical situation […] it is classified as class IIb”), we arrive at exactly the same table as in MDCG 2019-11 Rev.1.
According to this interpretation, the following classification of risk classes would be arrived at:
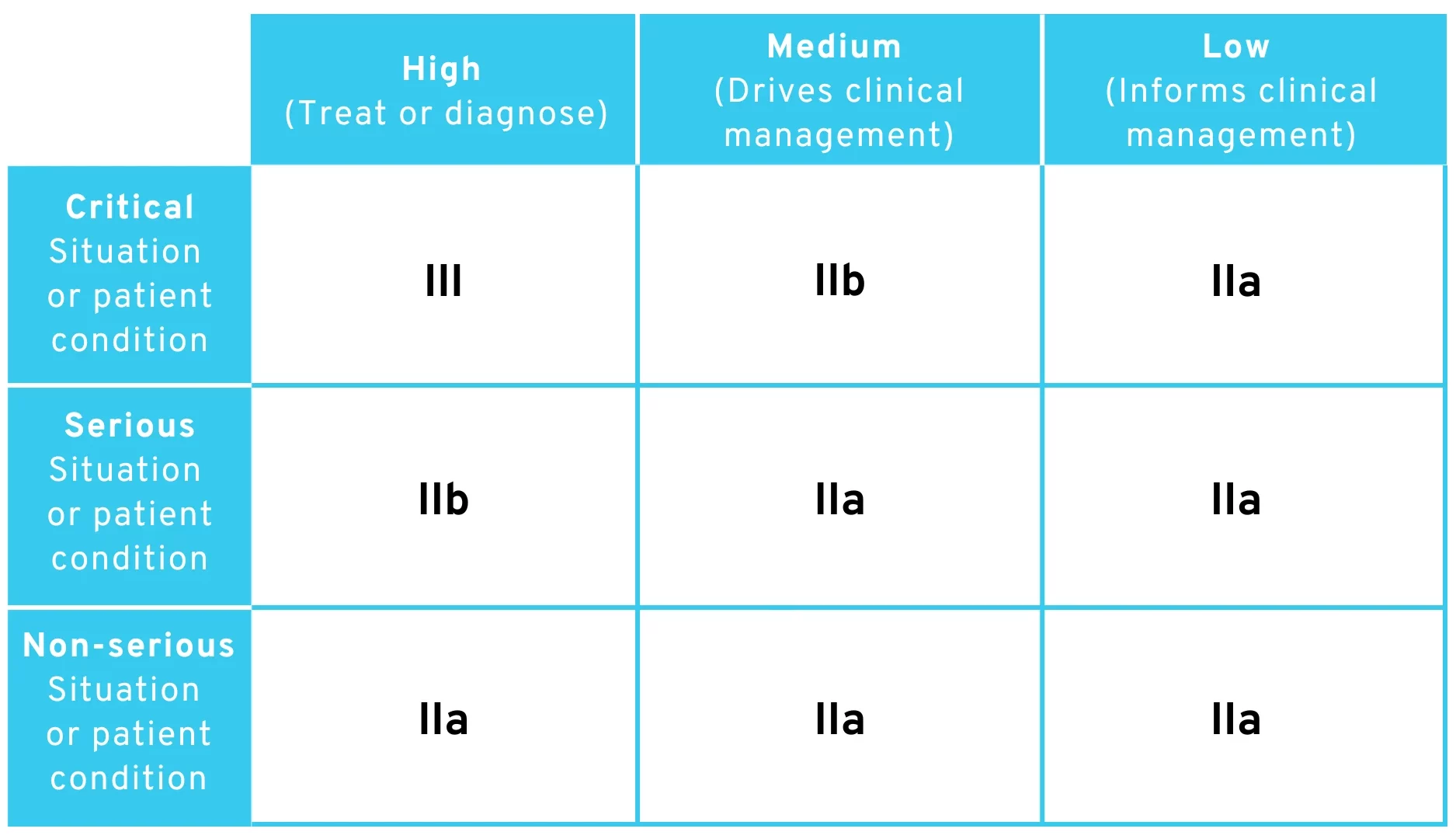
Proposed amendment to MDR Rule 11 – Table for classification of risk classes with literal interpretation
This interpretation is very likelybecause it would mean that the EU Commission had attempted to translate the table already published in the MDCG document into legal text. Interestingly, our team’s own analysis of the proposed Rule 11 amendment led us to exactly the same table in the MDCG document, without referring to it.
As you can see, risk class I is also missing from the classification here. It would therefore take some very creative reasoning to find ways to classify software as risk class I.
4. What else falls into Risk Class I with the Draft Amendments to the MDR?
The proposed amendment is particularly relevant for manufacturers of Class I products who manufacture products intended for use only by patients (medical laypersons). These include many digital health applications (DiGA) and other digital therapeutics (DTx).
The most popular argument currently used by these manufacturers targets a specific wording in the current MDR: “Software intended to provide information used to make decisions for diagnostic or therapeutic purposes belongs to class IIa, […] All other software is classified as class I.” Manufacturers argue that patients, as medical laypersons, cannot make diagnostic or therapeutic decisions. Read more about this in our article: Class I software according to the MDR – is that still possible?
However, this wording no longer appears in the amended Rule 11, which could make it more difficult for many manufacturers to justify risk class I. On the contrary, the use of MDCG terms in the legal text (e.g., “situation or patient condition” and “significance of information”) could also make their IMDRF definitions more relevant. There, laypersons are explicitly included as users of the medical device in the definition of a non-serious or serious situation. This means that applications for laypersons would also fall into at least Class IIa.
For us, it would be conceivable that this would shift the discussion about terminology to
- What does “clinical management” mean?
- What is a situation that is neither “critical,” “serious,” nor “non-serious”?
If you want to convince an examiner to classify your product as Class I in the future, you would therefore have to argue that either
- the significance of the information provided by the app is not even “low” or
- the patient’s condition is not even “non-serious.”
This argument would essentially add a column and row to the existing table:
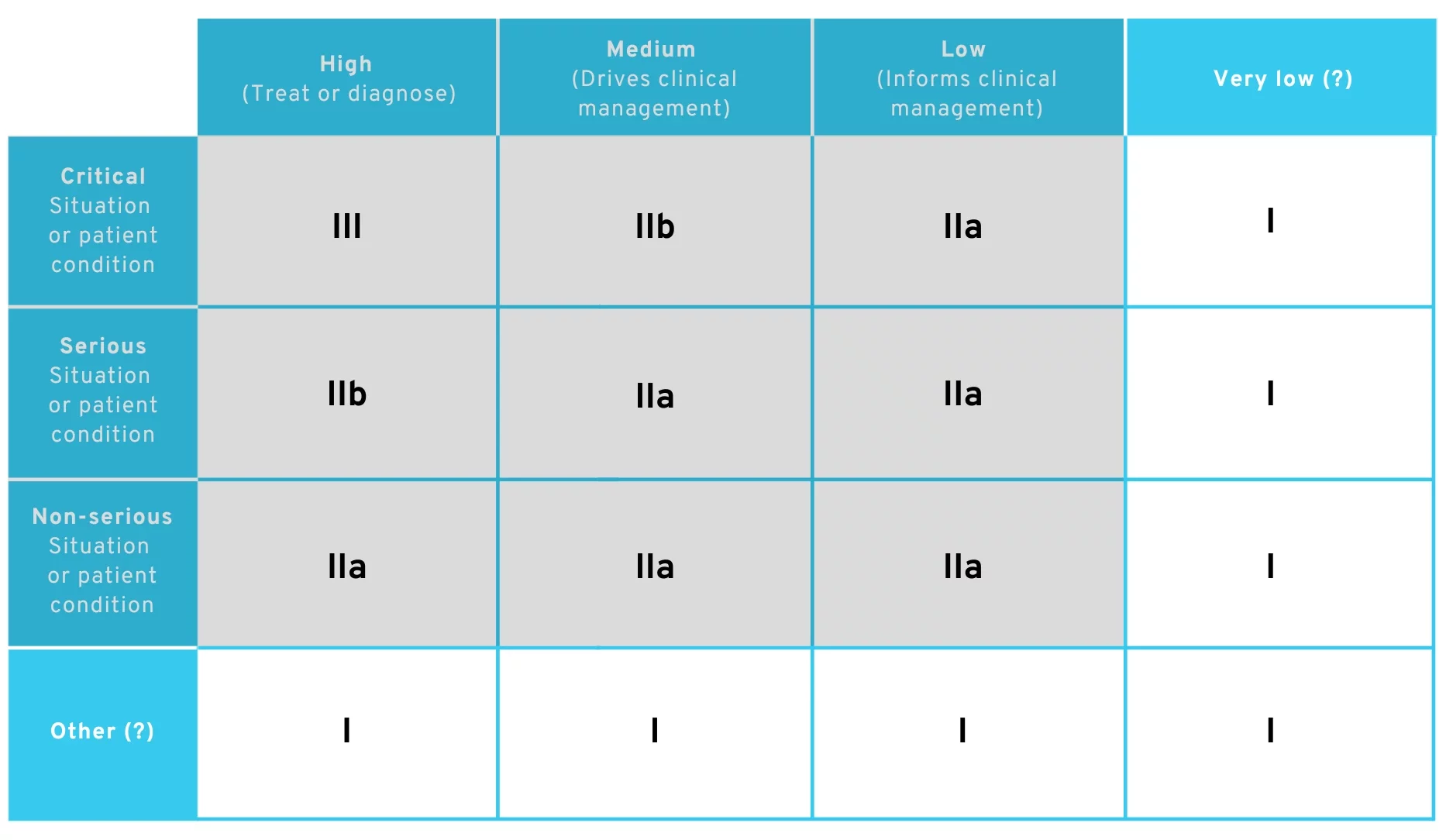
Proposed amendment to MDR Rule 11 – Possible routes in Class I
It is reasonable to argue that primary prevention applications continue to fall under Class I.. However, the ice is thin for any therapy-accompanying app, and it is not obvious how a Class I argument can still be achieved for this.
At least if the draft amendment to the MDR is actually implemented as it stands. However, this is far from certain, and adaptations are to be expected. This brings us to our next point:
5. Is this proposed change final and will it be implemented as such?
No. The proposed wording of the new Rule 11 is currently only a draft. It is an early version in the European legislative process. Experience shows that such drafts are subject to technical and political revisions as the process progresses. Changes are particularly likely in the case of complex issues such as software classification. We therefore do not currently expect Rule 11 to be incorporated into the final text of the law unchanged in its current form.
6. Timeline: When will the MDR changes and the new Rule 11 take effect?
It should be emphasized at this point that amending an EU regulation usually takes several years. We therefore expect a valid revision of the MDR (as of January 2026) to be available in 2027 at the earliest. This is also rather optimistic, and the process may take even longer.
Every proposal to amend an EU regulation undergoes a multi-stage process before being adopted:
- Legislative proposal: The European Commission drafts a formal regulation and submits it to the European Parliament and the Council of the EU for review.
- First reading: The European Parliament and the Council examine the proposal for the first time, can table amendments, and try to find a common position.
- Second reading: If no agreement is reached during the first reading, a second review by Parliament and the Council follows, during which further amendments may be made or the proposal may be rejected.
- Conciliation procedure: If Parliament and the Council still cannot reach agreement, a conciliation committee is set up to work out a joint compromise text within a fixed period.
- Third reading: The text negotiated in the conciliation procedure is finally adopted or rejected by both the European Parliament and the Council. Only then can the regulation enter into force.
The entire process is described in detail here.
We are closely monitoring further developments and will update this article on an ongoing basis. We will provide information about relevant changes in our newsletter.
7. Further Interpretations
There are other ways to interpret the proposed wording of Rule 11. However, the fact is that this wording will probably not end up in the MDR exactly as it stands after the relevant revisions. Accordingly, listing every possible interpretation, no matter how unlikely, is not expedient at this stage.
8. Conclusion
Based on the above interpretation, we consider the proposed amendment to Rule 11 to be serious. Under the new rule, most Class I software medical devices would very clearly fall into Class IIa (or higher), which would be a serious problem for many manufacturers. We currently have serious doubts as to whether this is intentional. It would completely contradict the stated goal of promoting innovation.
We also find the current wording of Rule 11 to be technically inconsistent. In particular, there is no clear and comprehensible representation of very low-risk software, as provided for, for example, in the 3×3 risk matrix proposed by the IMDRF. This matrix explicitly provides for a category for minor clinical effects, which would correspond to a Class I classification. This differentiation is lost in the current draft. We therefore assume that this is an unintentional error and will be corrected in future amendments to the draft.

Categories for Software as a Medical Device (SaMD) from the IMDRF
In general, we continue to look hopefully toward the future.The current Draft exists only in a first version. Several formal steps in the European legislative process still need to be taken before such an amendment to an EU regulation can be made.
Experience shows that the wording will be further adapted and clarified during this process. It is unlikely that the current draft of the MDR amendment will end up in the regulation in its final form.
We will keep you up to date on this in our newsletter.

