

With the EU AI Act, the EU is now also regulating the use of artificial intelligence (AI). This also affects manufacturers of software medical devices who integrate AI into their MDR medical devices (“Medical Device Artificial Intelligence” or MDAI). The AI Act is intended to ensure that AI systems are designed in such a way that the safety and fundamental rights of individuals are protected.
- But is the AI Regulation even relevant for my medical device?
- What additional requirements must medical device manufacturers in particular implement?
- And how do I manage to integrate these into my existing quality management system?
We answer these and other questions in this comprehensive guide to the EU AI Act. We focus entirely on the impact of the AI Regulation on MDR medical devices.
Contents of the guide to the AI Regulation
- 1. What is the AI Act? (Status 2026)
- 2. Is the AI Act relevant for my product?
- 3. The 3 product categories of the AI Act
- 4. Requirements for high-risk AI systems
- 5. How does the AI Act affect the approval of my medical device?
- 6. When will the AI Act come into effect? Implementation timeline
- 7. Conclusion
1. What is the EU AI Act?
The AI Act is a new regulation that aims to regulate the development and use of artificial intelligence (AI) in the EU. It is not limited to medical devices, but addresses any products that fall under the definition of an AI system (see next chapter, “Is the AI Act relevant to my product?”). The overarching goal of the AI Regulation is to protect the safety and fundamental rights of individuals. The complete regulation can be found on the website of the European Union.
2. Is the AI Act relevant for my product?
The AI Act defines AI systems as follows:
For the purposes of this Regulation, the term “AI system” means a machine-based system designed for varying degrees of autonomous operation, which may be adaptable after it has been put into operation and which, based on the inputs it receives, derives explicit or implicit objectives, such as outputs such as predictions, content, recommendations, or decisions that may influence physical or virtual environments.
In summary, an AI system is software that derives from the inputs it receives how a specific output can be generated. In addition, the system may (but does not have to) operate autonomously and continue to learn. The bottom line is that it is not yet 100% clear exactly which systems are excluded, but since most products with AI fall under machine learning, they are very clearly covered by this definition.
The MDCG has published a document that clarifies the interaction between the MDR and the AI Act: MDCG 2025-6. This document is of particular interest to manufacturers of software medical devices. It defines products that are subject to both the MDR and the AI Act as “Medical Device Artificial Intelligence (MDAI)”.
3. The 3 product categories of the AI Act
Do the same requirements apply to all AI medical devices (MDAI)? The short answer is no. Similar to the MDR, the EU AI Act also divides products into different classes:
- Prohibited AI practices
- High-risk AI systems
- Other AI systems (with low risk)
Which category does my AI or software medical device fall into? And what does that mean for me as a manufacturer? 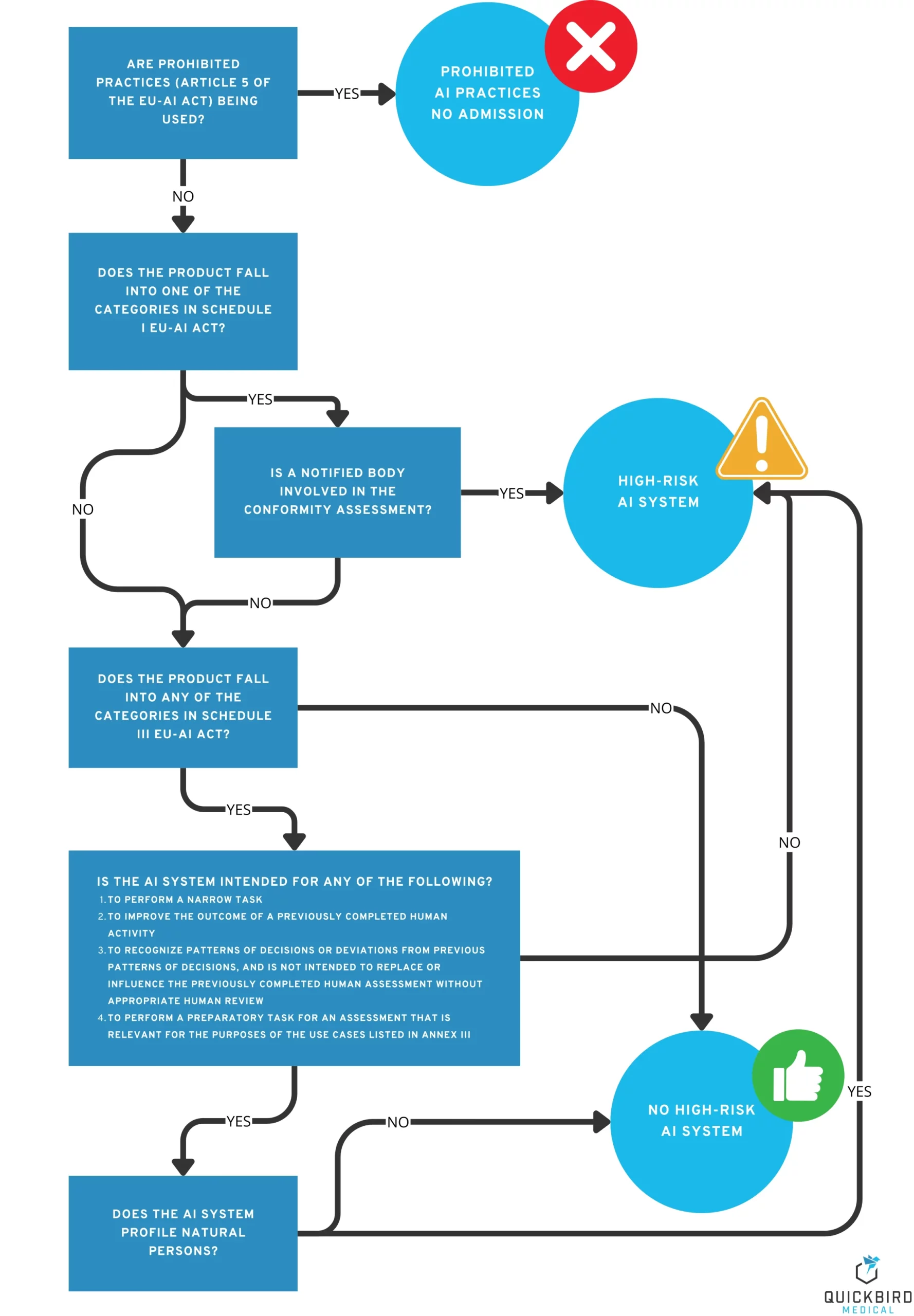
Decision tree: Which AI risk class does my AI or software medical device (MDAI) fall into?
3.1 Product category 1: Prohibited AI practices
Reduced to the essentials, it can be said that, above all, practices that aim to harm people – by subliminally influencing them or exploiting certain weaknesses or vulnerabilities (e.g. disabilities, age) – are prohibited. Article 5 of the AI Regulation lists a number of other prohibited practices, but these are generally not relevant to medical devices.
Does my medical device fall into this category? The goal of harming people usually contradicts the goals of a medical device, which is why you can generally assume that your AI does not fall into the category of “prohibited practices”. Nevertheless, you should take a close look at the criteria in Article 5 to be sure.
What requirements do I need to consider with regard to “prohibited AI practices”? The product may not be placed on the market.
3.2 Product category 2: High-risk AI systems
This category includes products that pose a high risk to the health and safety or fundamental rights of natural persons.
Does my medical device fall into this category? Especially when it comes to the terms “health” and “safety,” the idea of “medical devices” is not far off, especially since the MDR also places a strong focus on the safety of the products it regulates.
According to MDCG 2025-6, a medical device is a high-risk AI system if the following criteria are met:
- The AI system itself is a medical device or a safety component thereof.
- It is a notified body involved in conformity assessment according to MDR.
If a notified body is involved in the conformity assessment procedure for your product (e.g., because it falls into risk class IIa or higher), your medical device is automatically a high-risk AI system according to the AI Regulation (see decision tree in the figure above). The notified body must then check not only the requirements of the MDR, but also those of the AI Act as part of the conformity assessment. However, if no notified body is involved in the approval of your product (e.g., because the product falls into risk class I according to the MDR, or it is not a medical device at all), you must check Annex III of the AI Act in particular. This contains a list of systems that are considered high-risk AI systems. However, most medical devices will not be found on this list, which is why we assume that many medical devices in risk class I according to the MDR are not high-risk AI systems.
This assessment is also confirmed by MDCG 2025-6, which contains the table below:
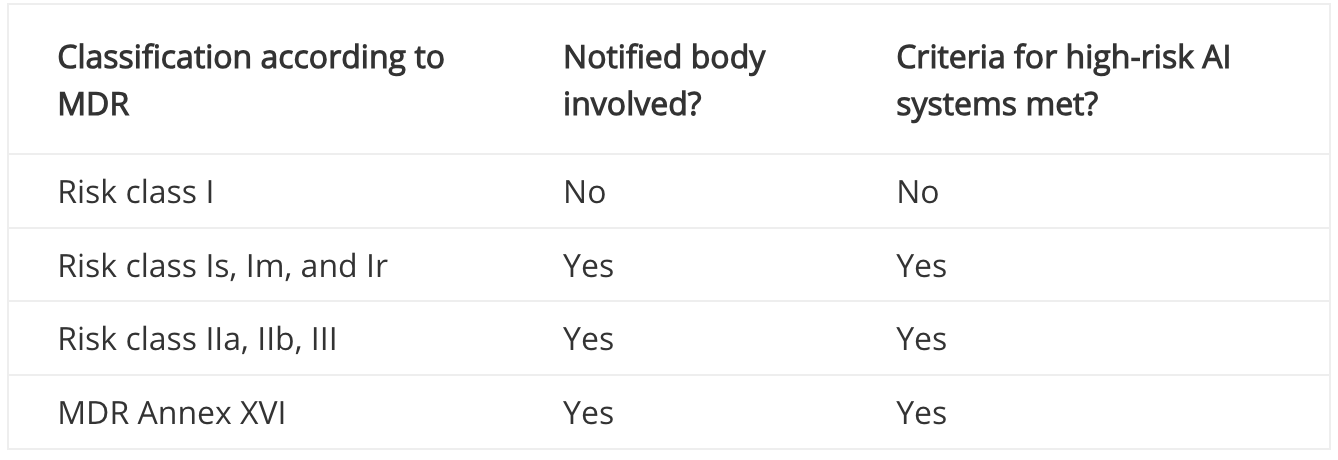
Classification of high-risk AI systems according to MDR
What requirements do I need to observe? If your product falls into the category of high-risk AI systems, the AI Regulation may have a significant impact on your organization and your medical device. This is because the contents of the EU AI Act specifically define requirements for the manufacturers of such systems and the systems themselves. We provide an overview of the requirements below in the chapter “Requirements for high-risk AI systems”.
3.3 Product category 3: Other AI systems (low risk)
All AI products that cannot be classified as either prohibited AI practices or high-risk AI systems fall into this category. Roughly speaking, these are systems that do not pose any significant risks to the safety and fundamental rights of individuals. Does my medical device fall into this category? If your product fulfills the following criteria, it probably falls into this category::
- It is a class I risk product according to MDR (no notified body is involved in the conformity assessment).
- It was ruled out that it is a high-risk AI system.
- It was ruled out that these were forbidden AI practices.handelt
What requirements do I need to observe? Compared to high-risk AI systems, products in this category come off really well. In fact, the entire AI Act contains only one requirement for such systems: transparency obligations. And these only apply to systems that interact with humans, generate media content, or are used for emotion recognition. Roughly speaking, manufacturers of such products must ensure that users know that they are dealing with AI and that the content generated is not real images, videos, etc. (e.g. deepfake). AI systems in this category do not have to be registered in a separate database and they do not require a declaration of conformity. So if your medical device falls into this category, the effort required for compliance with the AI Act is likely to be manageable for you.
3.4 Additional category: AI models for general use
Regardless of their risk classification, certain AI models can be considered “general-purpose AI models.” These are models that can be used for various purposes.
However, since the purpose of medical devices is usually very precisely defined, this category will only be relevant for a few manufacturers in practice. For the sake of completeness, we would nevertheless like to mention it here.
If you are developing an AI model that goes beyond application in a specific product, you should take a closer look at this category.
The exact requirements for such models are defined in Chapter V of the AI Act.
4. AI Regulation – Requirements for high-risk AI systems and their manufacturers
The exact requirements for high-risk AI systems and their manufacturers are specified in Chapter III, Section 2 and Section 3 of the AI Act. At this point, we assume that you already meet the requirements of the MDR. Thus, we will focus here on the points that go beyond the requirements of the MDR. Learn more about the MDR requirements for AI medical devices in this article: Approval & Certification of Software Medical Devices (MDR)
The AI Regulation defines the obligations of providers of high-risk AI systems as follows (Article 16). You have to …
a) ensure that their high-risk AI-systems meet the requirements set out in Section 2;
(b) indicate their name, registered trade name or registered trade mark and their contact address on the high-risk AI system or, where that is not possible, on its packaging or in the documentation accompanying it;
(c) have a quality management system that complies with Article 17;
(d) keep the documentation referred to in Article 18;
(e) keep the logs automatically generated by their high-risk AI-systems in accordance with Article 19, if they are under their control;
(f) ensure that the high-risk AI-system is subject to the relevant conformity assessment procedure referred to in Article 43 before being placed on the market or put into service;
(g) draw up an EU declaration of conformity in accordance with Article 47;
(h) affix the CE marking to the high-risk AI system, or, where that is not possible, on its packaging or in the documentation accompanying it, to indicate conformity with this Regulation in accordance with Article 48;
(i) comply with the registration obligations referred to in Article 49(1);
(j) take the necessary corrective measures and provide the information required in accordance with Article 20;
k) upon reasoned request by a competent national authority, demonstrate that the high-risk AI system meets the requirements in Section 2;
l) ensure that the high-risk AI system meets the accessibility requirements in accordance with Directives (EU) 2016/2102 and (EU) 2019/882.
Don’t worry, we won’t leave you alone with this list. In the following, we would therefore like to shed some light on what this means for you as a manufacturer of an AI medical device. We will address the most important points that lead to an adaptation of your management system and/or medical device.
The requirements of the EU-AI Act and the MDR overlap in many respects.
4.1 AI Act – Requirements for your quality management system
The MDR already requires medical device manufacturers to establish a quality management system. In most cases, ISO 13485 and – when software is involved – IEC 62304 are implemented, among others. At first glance, it may appear that you have also fulfilled the requirements of the AI Act. On closer inspection, however, it becomes clear that this is only partially true. But don’t worry, the majority of your system can remain as it is. You just need to make a few adjustments and additions. The requirements from Article 17 of the EU AI Act are listed in the table below. In addition to the exact requirements of the regulation, we have defined the specific measures for you that you must implement to ensure the corresponding conformity of your quality management system. The quality management system must include at least the following aspects:
(Note: Our comments are not meant to be absolute truths, but merely to highlight any similarities and differences between the AI Act and your existing system. Therefore, for each point, check to see how you need to take action.)
| Wording from the AI Act | commentary on implementation |
|---|---|
| a) a concept for regulatory compliance, which includes compliance with the conformity assessment procedures and the procedures for managing changes to the high-risk AI system; | Since you, as a medical device manufacturer, already have a concept for compliance with the MDR, GDPR and other regulatory requirements, you should simply extend this to include the AI Act. |
| b) techniques, processes and systematic measures for the design, design control and design review of the high-risk AI system; | This point could already be implemented by ISO 13485 and IEC 62304. |
| c) Techniques, procedures and systematic measures for the development, quality control and quality assurance of the high-risk AI system; | this point should already be implemented by ISO 13485 and IEC 62304. |
| (d) the investigation, testing and validation procedures to be carried out before, during and after the development of the high-risk AI system and the frequency of implementation; | This topic can also be found in ISO 13485 and, as a medical device manufacturer, you already have corresponding processes in place. However, check whether your procedures are appropriate for the AI system. |
| e) the technical specifications and standards to be applied and, where the relevant standards are not applied in full or do not cover all relevant requirements in accordance with Section 2, the means by which it is intended to ensure that the high-risk AI system meets those requirements; | However, harmonized standards for implementing the AI Act will be established by the European Commission over time (as of January 20, 2026). This will probably be similar to the MDR, whose harmonized standards are published in an implementing decision and are continuously being expanded. |
| f) systems and procedures for data management, including data extraction, data collection, data analysis, data labeling, data storage, data filtering, data interpretation, data aggregation, data retention and other operations performed on data in advance of and for the purpose of placing on the market or putting into service high-risk AI-systems; | These procedures will probably have to be newly established, as they are not explicitly required by the MDR or the associated standards. You can find out more about this in the “Data and data governance” section below. |
| (g) the risk management system referred to in Article 9; | This point should be largely covered by the MDR (or ISO 14971). Check to what extent your risk management already meets the requirements of the AI Act (see following chapters). |
| h) the establishment, implementation and maintenance of a post-market surveillance system in accordance with Article 72; | This point is already largely covered by the MDR (post-market surveillance). Check to what extent your post-market surveillance system already meets the requirements of the AI Act (see following chapters). |
| (i) the procedure for reporting a serious incident as referred to in Article 73; | This point is largely covered by the MDR (vigilance). There is even a special regulation for medical devices. Since medical device manufacturers already have such a system in place, the AI Regulation merely requires that the new requirements be integrated into this system. You will need to assess the extent to which these requirements will result in changes to your system. It may well be that little or no effort is required here. |
| (j) the management of communication with national competent authorities, other relevant authorities, including authorities granting or facilitating access to data, notified bodies, other stakeholders, customers or other interested parties; | Communication with various stakeholders must already be regulated by ISO 13485. So simply add any new authorities or stakeholders to this list to achieve conformity with this point. This could become relevant, for example, if you want to use data from an AI real-world laboratory. |
| (k) systems and procedures for recording all relevant documentation and information; | You should have already implemented this point through your quality management system. The aim here is to establish processes that generate the necessary outputs (e.g. technical documentation). |
| l) Resource management, including measures with regard to security of supply; | You should have already implemented this point through your quality management system in accordance with ISO 13485. |
| m) an accountability framework that governs the responsibilities of management and other personnel with regard to all aspects listed in this paragraph. | You can transfer this responsibility to your PRRC according to MDR, or define a new role for it. |
4.1.1 Risk management
As a medical device manufacturer, you already have a risk management system in place and may be subject to various laws governing risk management activities (e.g., MDR, GDPR). However, the requirements for the risk management system are not identical in the MDR and AI Act! Probably the most fundamental difference is that you must consider not only risks relating to safety, but also to the fundamental rights of individuals. In some cases, this can be implemented by complying with other regulations (e.g., GDPR), but in addition to physical integrity and the protection of personal data, other rights may also be violated by an AI system. Be sure to take a look at the Charter of Fundamental Rights of the European Union. In addition, high-risk AI systems must be regularly tested using appropriate procedures (this presumably applies above all to continuously learning systems, which can only be approved in exceptional cases under the MDR). This testing is also necessary to determine the most appropriate risk management measures. The identified risks are reduced to the extent technically possible and to a justifiable degree.
4.1.2 Post-market surveillance system
The MDR already requires you to ensure the post-market surveillance of your medical device. This is also explicitly addressed in the AI Regulation. If you have already established such a system under the MDR, you must check whether it also fulfills the following requirement: the system must enable you to continuously assess the conformity of the product.
4.1.3 System for reporting serious incidents and malfunctions
Due to your already established vigilance system in accordance with the MDR, there is little to implement in this area. The AI Act even has a special rule for medical devices. As there is already an obligation to report safety-related incidents, you only need to expand your system to include the reporting of “breaches of the provisions of Union law on the protection of fundamental rights”.
4.1.4 Data and data governance
As a manufacturer, you are obliged to establish suitable processes that regulate the following:
- Conceptual decisions (e.g. choice of model type, definition of relevant features)
- Data collection (e.g. data sources)
- Data preparation processes (e.g. labeling, cleansing, aggregation)
- List of relevant assumptions (e.g. relationship between features, potential influencing factors)
- Prior assessment of the availability, quantity and suitability of the required data sets
- Investigation of possible distortions/bias and appropriate measures to detect, prevent and mitigate them
- Identification of possible data gaps, deficiencies and measures to rectify these
In principle, this chapter also places specific requirements on your training, validation and test data. You should integrate these requirements directly into your processes. Tip: When creating these processes, also consider the requirements for technical documentation in Annex IV and Article 11 of the AI Regulation. This will ensure that your procedures also generate the necessary outputs.
4.1.5 Retention requirements
If you comply with the MDR’s retention requirements, you probably also fulfill those of the AI Act. This defines that you must retain your documentation for a period of 10 years after the AI system has been placed on the market. According to Article 18 of the AI Regulation, this documentation includes the technical documentation, the quality management system, changes released by notified bodies and documents issued, as well as the declaration of conformity. Protocols automatically generated by the product must be kept for 6 months.
4.2 AI Act – Requirements for technical documentation
“Technical documentation” is a term that you are probably already familiar with from the MDR. You have to create technical documentation for your medical device anyway. In fact, there are many parallels here too, and you will have already implemented most of the points by complying with the MDR. These include, for example, the product description, a declaration of conformity and the methods used in product development. However, this does not apply in every case! In particular, software systems in software safety class A according to IEC 62304 may have some special features with regard to the technical documentation. The AI Regulation requires you, for example, to document a system architecture and the definition of interfaces to other systems. This goes beyond the requirements of IEC 62304 for safety class A software systems. Tip: It is therefore not possible to say in general terms to what extent your technical documentation needs to be expanded. It is best to go through Annex IV of the AI Act and check for each point to what extent you have already implemented it in your current documentation. Ideally, you should then create any missing information as part of the newly defined or adapted processes.
4.3 Product requirements
4.3.1 Recording and logs
High-risk AI systems are required to keep logs. This is therefore a clear technical requirement. As is often the case, these logs must be kept to an extent that is “adequate for the purpose”. Article 12 of the AI Regulation defines what this means. According to this article, the logs must, above all, record events that are relevant for the following: (a) the identification of situations that could result in the high-risk AI-system posing a risk as referred to in Article 79(1) or in a significant change occurring; b) facilitating post-market monitoring in accordance with Article 72; and c) monitoring the operation of high-risk AI systems in accordance with Article 26(5). Essentially, the aim is to monitor and trace situations associated with risks and damage as effectively as possible. When implementing this logging, manufacturers must adhere to recognized standards and specifications (e.g., BSI requirements).
4.3.2 Transparency and provision of information to users
If you don’t yet have instructions for use for your medical device, you need them now at the latest. If you already have instructions for use, you will probably need to revise them. Even though some requirements are already covered by the MDR, the AI Act now also requires information on aspects such as cybersecurity and risks to fundamental rights. The degrees of accuracy and relevant accuracy indicators of the system must also be stated here. So check your current instructions for use to see how you are already fulfilling the obligations of the AI Act, and then make the necessary adjustments.
4.3.3 Human supervision
The duty of human supervision is a mandatory risk control measure for high-risk AI systems. The system must be designed and developed in such a way that it can be effectively supervised by a human. Here, some parallels can be seen with the transparency requirements. Essentially, it must be possible for a user to understand what is happening in the product and what it is outputting. They must also be able to shut down the system or intervene in its operation. The option for supervision can either be built directly into the software or handed over to the user. Roughly summarized, it must simply be possible to detect anomalies or misconduct and to stop the system in case of doubt.
4.3.4 Accuracy, robustness and cybersecurity
High-risk AI-systems must be resilient to errors, faults or discrepancies that may occur within the system or the environment in which the system operates, in particular as a result of its interaction with natural persons or other systems. The robustness of high-risk AI-systems can be achieved through technical redundancy, which may include back-up or fault tolerance plans. High-risk AI systems that continue to learn after being placed on the market (or put into operation) are to be developed in such a way that appropriate risk-mitigation measures are taken to address any potentially biased results. High-risk AI systems must be resilient to attempts by unauthorized third parties to alter their use or performance by exploiting system vulnerabilities. The technical solutions for ensuring the cybersecurity of high-risk AI systems must be appropriate to the respective circumstances and risks. Technical solutions for dealing with AI-specific vulnerabilities include, where appropriate, measures to prevent and control attacks that attempt to manipulate the training data set (“data poisoning”), input data that is intended to trick the model into making mistakes (“adversarial examples”), or model deficiencies.
4.3.5 CE marking
Your medical device already has a CE marking if you have approved it in compliance with the MDR. You just need to reference the appropriate regulation so that users can see that your product has been developed in accordance with both regulations (MDR and AI Act).
4.3.6 Registration of the product
Similar to EUDAMED for medical devices, there will also be an EU database for high-risk AI systems in the future, in which your product must be registered. However, this database is not yet publicly accessible (as of January 20, 2026).
5. How does the AI Act affect the approval of my medical device?
The risk class of your medical device according to MDR affects the applicability of the AI Act. You can find out how to determine the risk class according to MDR in our guide to classifying software medical devices.
5.1 Risk class I
The AI Act does not change anything with regard to the approval of Class I products according to MDR (except for the requirements in Chapter 3.3) – as long as the product is not a high-risk AI system. However, if this is the case, you will need a notified body in the future to assess conformity with the AI Regulation. Be sure to keep this in mind when planning to implement a risk class I product, as it can significantly delay market entry.
Please note: Exceptions apply to products in risk classes Im, Is, and Ir, for which a notified body is involved in the conformity assessment. However, these are rather rare in the case of pure software products.
5.2 Risk class IIa or higher
Products in risk class IIa or higher are basically automatically classified as high-risk AI systems under the AI Act. This means that a number of additional requirements must be implemented, as already outlined above in this article. When it comes to approval (especially conformity assessment), you must ensure that your notified body is also accredited under the AI Regulation or is seeking such accreditation. As part of the conformity assessment, the notified body must then verify not only conformity with the MDR, but also with the AI Act.
However, the conformity assessment is not expected to be carried out separately for both regulations, but rather as part of an integrated assessment.
Only if the product complies with both regulations may it bear the CE symbol. Therefore, contact your notified body as soon as possible to clarify whether such accreditation is planned for the AI Regulation.
In June 2025, the MDCG published a document describing the interaction between the MDR and the AI Act: MDCG 2025-6.
6. When will the AI Act come into effect? Implementation timeline
The EU AI Act will not come into force immediately in its entirety, but rather over time. The current timeline stipulates that the majority of the regulation will come into force by August 2, 2026, and that it will be fully valid from August 2, 2027.
This is evident from the European Commission’s timeline:
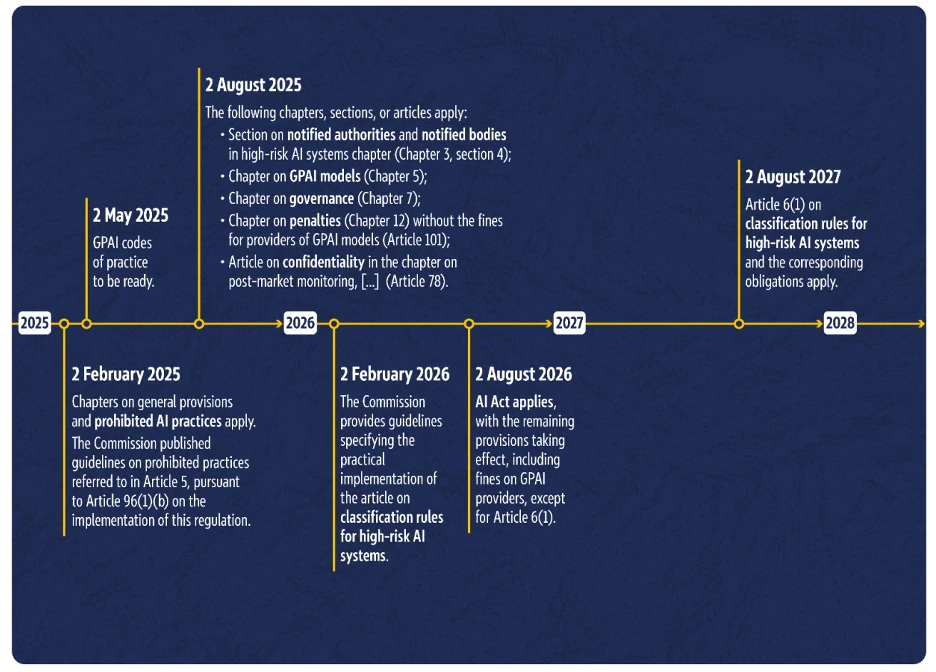
Implementation timeline for the European Commission’s AI Act (Source: AI Act implementation timeline)
In summary, this means the following for manufacturers of AI and software medical devices (MDAI):
- Many fundamental rules of the AI Act are already in effect today (e.g., prohibited AI practices).
- From August 2026, almost the entire AI Act will apply, with one important exception: Article 6(1). This describes the classification rules for high-risk AI systems.
- Manufacturers of high-risk AI systems therefore have until August 2027 to implement all requirements for their product and for themselves as manufacturers (unless it is a system listed in Annex III). This is because the classification as a high-risk AI system will only apply from this point in time.
7. Conclusion
The AI Act differs from the MDR primarily in its objectives. While the MDR aims primarily to ensure the safety and functionality of medical devices, the AI Regulation also includes the protection of individuals’ fundamental rights. Basically, it can be said that many new requirements will apply primarily to MDR medical devices in risk class IIa or higher. These are automatically assigned to the category “high-risk AI systems” according to the AI Act. However, medical devices in risk class I may also fall into this category in individual cases. For products that are not high-risk AI systems and do not fall under the “prohibited practices”, the AI Regulation brings few changes. Manufacturers of high-risk AI medical devices must make adjustments in a number of areas in order to comply with the requirements of the AI Act. In addition to the requirements of the AI Act, you must of course also comply with the requirements of the MDR that are relevant to artificial intelligence.
Do you need support with the development of a medical AI system? We develop medical AI systems for companies in the healthcare sector on a contract basis.
Our AI services: – Technical review of medical AI systems – Development of new machine learning models in the healthcare sector – Individual advice on regulatory compliance for your planned AI-based product
We help you to implement the extensive requirements of the MDR and the AI Act and develop a successful product.

