

Are there still Class I software medical devices according to MDR? Are regulatory authorities really withdrawing Class I software products from the market in droves? Which federal state do I need to go to in order to remain on the market with my Class I software?
As rumors have been circulating recently that it is no longer possible to obtain approval for software medical devices in risk class I, we would like to shed some light on the matter with this article.
This article is primarily aimed at manufacturers of medical device software that poses little or no risk. This includes, for example, training apps that are used autonomously by patients and cannot cause any harm to them.
Update – June 2025: The guidance document “MDCG 2019-11: Guidance on Qualification and Classification of Software […]” has been available in its revised version 1 since June 2025. This article takes all of the changes into account. A detailed overview of the specific changes can also be found here.
Update – December 2025: In December 2025, the EU Commission published a draft amendment to the MDR, which includes an adjustment to Rule 11 on the classification of software medical devices. We discuss the possible implications for risk class I software in Chapter 3 of this article.
Table of Contents
- 1. MDR risk classes
- 2. Update of the official MDCG Guidance (Revision 1) – June 2025
- 3. Draft Amendment to the MDR & Implications for Class I Software – December 2025
- 4. Dealing with uncertainty in the risk class
- 5. Why we urgently need risk class I in the EU
- 6. Conclusion
1. Risk classes for medical devices under the MDR
As we have already described the risk classes of the MDR in detail in our software classification guide, we would like to keep it brief at this point:
- The MDR essentially distinguishes between risk classes I, IIa, IIb and III for software
- The decisive factor in determining the risk class of a product is its intended purpose
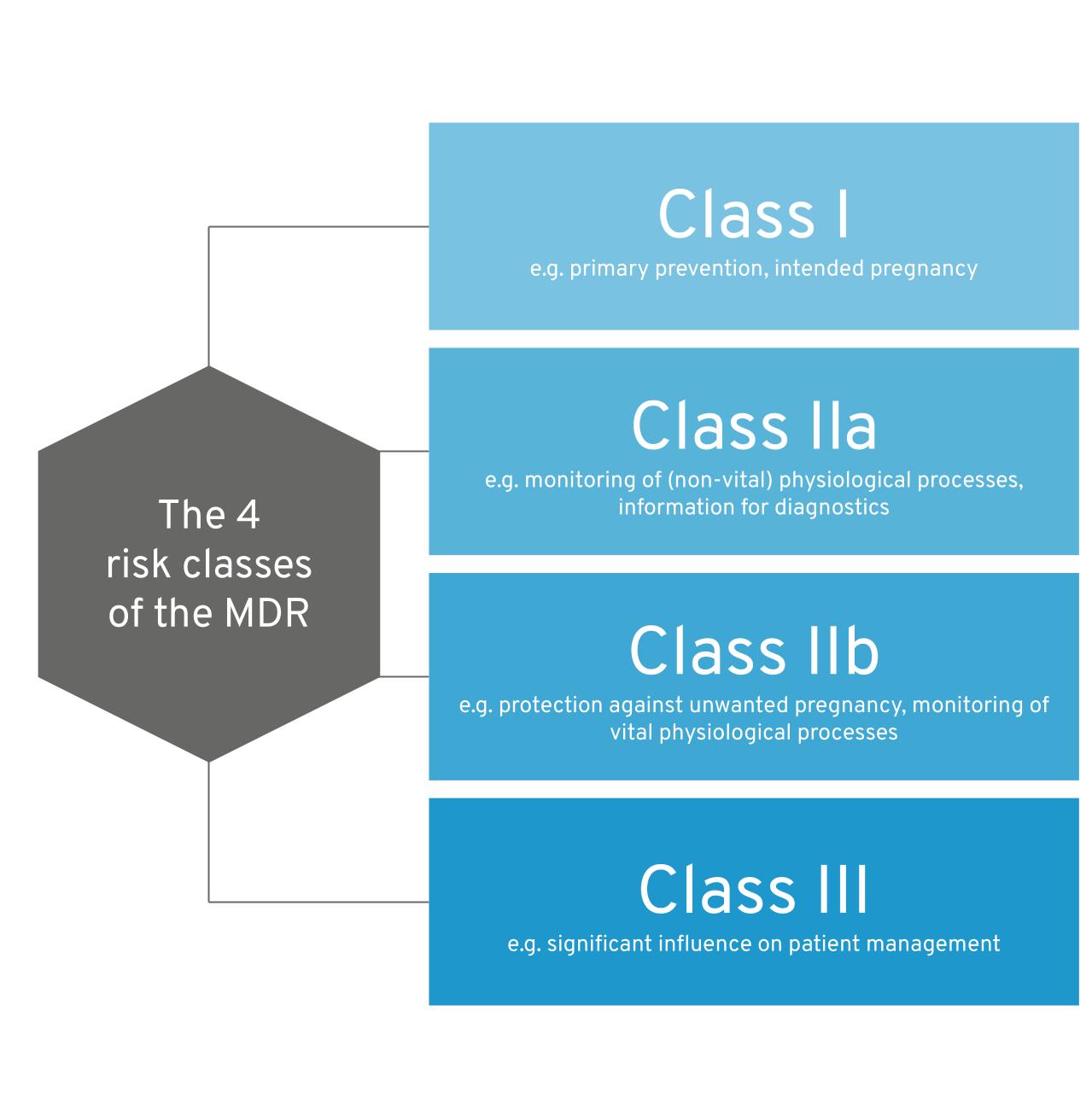
The 4 MDR risk classes at a glance
If you would like to find out more about the risk classification of software medical devices, the following guidelines from us will help you:
- Qualification of software medical devices – Guidelines: Is your app a medical device?
- Classification of software medical devices – guide: Which risk class do you fall into?
Compared to higher-class products, the approval of Class I risk products is much more resource-efficient and can be completed more quickly. Many manufacturers therefore ask themselves whether their product is a Class I or Class IIa product.
You will find out why this is the case in the next chapter.
1.1 Risk class I or IIa – Why is this important?
Why is the distinction between Class I and IIa so important? And why is there so much discussion about it?
Risk class I products differ significantly from higher-class products.
Class I products …
- do not require a certificate from a notified body for market approval,
- can enter the market without delay (caused by an audit) and do not have to bear the costs of a notified body,
- are only subject to unannounced inspections by the respective supervisory authority of the federal state. These are significantly less costly than certification by a notified body (risk class IIa and higher) and are not subject to a fee.
To illustrate this even better, we would like to show you some empirical values here. A class IIa product means compared to class I:
- Delay in market approval by 6-18 months (duration of the conformity assessment procedure of a notified body and issuance of the CE certificate)
- Costs of up to 6-digit euro amounts for the conformity assessment procedure and certification of the QMS
- Obligation to notify significant changes and further testing by the notified body for product changes after approval
The time required to successfully complete a conformity assessment procedure varies greatly and depends on a number of factors. In any case, this is another unknown factor that is difficult to factor into product planning.
In many cases, the distinction between risk classes I and IIa therefore really determines the existence of products and entire companies.
That is why Class I is particularly interesting for start-ups that are dependent on rapid market approval and have limited resources.
You can find out exactly what to expect with a risk class IIa product compared to class I in a separate technical article we have published: MDR risk class I vs. IIa: Differences for software medical devices
1.2 MDR – Rule 11 – Clear distinction between Class I and IIa?
The answer to whether your software or app can fall into risk class I can be found in the Medical Device Regulation (MDR) – Rule 11.
Quote from MDR 6.3 Rule 11 (abbreviated):
Software intended to provide information used to make decisions for diagnostic or therapeutic purposes is in class IIa,
[…]
All other software is assigned to class I.
Many manufacturers are asking themselves this question:
-
- What is information that is used to make decisions for diagnostic and therapeutic purposes?
- Who can “use” such information to make diagnostic or therapeutic decisions?
- Can an app that only provides a patient with training modules and knowledge influence therapy or diagnostics at all?
- Is the recommendation to do a certain exercise already a therapeutic decision?
Surprise: There are various ways of interpreting this Rule 11. This is because the BfArM, notified bodies and manufacturers do not agree on this. There is a lot of room for interpretation, which is why there are always discussions and rumors. We would therefore like to describe the 2 most important interpretations of this Rule 11 here.
1.2.1 Interpretation 1: All software provides information that influences therapy or diagnostics.
According to this interpretation, an app for patients also provides information for decisions on the treatment or diagnosis of diseases. The patient would be treating or diagnosing themselves, and thus sentence 1 of rule 11 would apply.
Strictly speaking, this interpretation means that there is no such thing as Class I standalone software, as software by definition always generates output (information) based on input. One exception would be, for example, an app that displays exactly the same content to every user without any individualization. However, this raises the question of whether it is actually a medical device (see also our guide “Is your app a medical device?”).
1.2.2 Interpretation 2: Only healthcare professionals can make therapeutic or diagnostic decisions
This is the manufacturer-friendly interpretation of Rule 11. According to this interpretation, a training app that is only used by patients would fall into risk class I. The reasoning behind this is that a layperson is not in a position to make therapeutic or diagnostic decisions. Sentence 1 of Rule 11 therefore does not apply – information is not used for decisions for diagnostic or therapeutic purposes.
Note: However, the situation is different for products that also involve medical professionals in their use and provide supporting information (e.g., via a web dashboard). This was decided, for example, by the Hanseatic Higher Regional Court in the Dermanostic case. In this case, a competitor initiated legal proceedings against Dermanostic. The court then ruled that Dermanostic was a Class IIa product and not a Class I product. You can read more about this here.
1.2.3 Whose interpretation counts?
We can see that different stakeholders have different views:
-
- BfArM
- State supervisory authority
- Notified body
- Manufacturer
- Court
If you decide to go down the route of a risk class I product, your own opinion (as the manufacturer) is particularly relevant, but of course the opinion of your state supervisory authority is also important. This authority is primarily responsible for monitoring manufacturers in your region.
Court rulings are also important, of course, in the event of a legal dispute (e.g., between you and a competitor) – see Dermanostic.
The assessment of notified bodies is particularly important if you are aiming for risk class IIa or higher. For risk class I products, these are not involved at all.
The BfArM primarily has an indirect influence on risk classification. As it is a federal authority, the BfArM can also exert influence on state supervisory authorities and, for example, issue recommendations that state authorities may follow.
1.3 Conclusion – is Class I still viable?
Yes, it is still possible to obtain approval for Class I software medical devices. However, this depends heavily on your state regulatory authority. Under certain circumstances, it may be worthwhile to relocate your company’s headquarters to a state whose regulatory authorities are “Class I-friendly.”
Alternatively, we offer another exciting model. You can commission us as an external distributor to bring your medical device to market. We offer this service for software-based medical devices and DiGA: More information
Please feel free to contact us if you need an external distributor for your app or other standalone software.
2. Update of the official MDCG Guidance (Revision 1) – June 2025
The official MDCG guidance on the qualification and classification of medical device software (MDSW) was updated in June 2025 in Revision 1. The update was long awaited and caused considerable uncertainty in the digital health scene. The key question is: Does the new version spell the end of risk class I software?
Fortunately, this is not the case. In fact, little has changed with regard to the (sometimes vague) recommendations for risk classes I and IIa. We have compiled a detailed overview of all changes here: All changes to MDCG 2019-11 Rev.1
3. Draft Amendment to the MDR & Implications for Class I Software – December 2025
On December 16, 2025, the EU Commission published a draft amendment to the Medical Device Regulation (MDR). A central component of this draft is a fundamental revision of Rule 11, which we have explained above in its currently valid version.
At first glance, the new wording seems promising for manufacturers: the logic of classification appears to be reversed, so that software initially falls into Class I unless higher classification criteria apply. However, closer analysis shows that this hope could be deceptive. Depending on the interpretation, the draft would result in the majority of medical software being classified at least in Class IIa.
We have analyzed the draft amendment, the new Rule 11, and its possible interpretations in detail and discussed the specific implications for Class I software in a separate technical article: To the technical article
It is important to note that the published text is an early draft in the European legislative process. Experience shows that the wording will be further amended in the coming years. It is therefore unlikely that this draft for Rule 11 will be incorporated unchanged into the final MDR text.
4. Dealing with uncertainty in the risk class
How can I be sure that my risk class is correct? There is never absolute legal certainty in this area. In principle, a higher classification is always “safer,” but also much more complex and expensive.
However, since many companies are unwilling or unable to afford such a procedure, we would like to outline a few options available to you as a manufacturer of a Class I product.
- Read the MDCG Guidance documents (especially MDCG 2019-11).
- Look at similar products on the market that are approved under MDR. In particular, you should consider products from manufacturers for which your supervisory authority is also responsible.
- Get expert advice. There are many consultants who have been involved in the approval of many Class I medical devices. In many cases, this experience can be transferred to your product. Feel free to contact us if you need support.
- Obtain a legal opinion. This is issued by a lawyer and can support your arguments before a supervisory authority, but also in court. It creates additional persuasive power. Please note, however, that such an expert opinion cannot provide absolute legal certainty. Contact us if you are interested in an expert opinion. We will be happy to put you in touch with a lawyer who is familiar with the subject.
4.1 What happens if I register my product under risk class I even though I am unsure?
In principle, you can do that, of course. It is the manufacturer’s responsibility to classify their products correctly.
BUT be aware that this may pose a business risk. Some of these risks include:
| Scenario | Hazard | Damage |
| The product really does harbor risks in use | A patient could injure themselves | Legal consequences, reputational damage |
| A competitor initiates legal proceedings against you | The court decides that you must classify your product higher | Product must be withdrawn from the market or classified higher |
| A regulator reviews your product and disagrees with the risk classification | The supervisory authority disagrees with the risk class of your product | Product must be withdrawn from the market or classified higher |
5. Why we urgently need risk class I in the EU
There are countless innovative software products with medical applications that pose virtually no risk to patients (e.g., training apps for reducing anxiety). Access to such products has been greatly promoted in recent years (e.g., through the introduction of DiGA), and thousands of patients use such products effectively every day to improve their health. This not only relieves the burden on patients, but also on the entire healthcare system.
In our view, the effort involved in obtaining approval under risk class IIa is disproportionate to the generally very low risks posed by patient-focused lay products.
Such products often originate from the startup environment, which usually has to make do with limited resources. Overregulation (and, to put it bluntly, making Class I software impossible) stifles such innovations in their infancy. Small companies simply cannot afford to go through a conformity assessment procedure with a notified body—neither in terms of time nor money.
This is not only detrimental to Germany as a business location, as manufacturers relocate their sites to other countries, but ultimately also prevents better patient care from a political perspective.
We therefore urgently need to retain the ability to develop and approve Risk Class I software.
6. Conclusion on risk class I according to MDR
After all the rumors and discussions, the question remained: “Are there still standalone software medical devices in risk class I?”
Although the situation is still not fully clarified after extensive research and numerous discussions within our circle, some insights can be gained:
-
- Regulatory authorities, notified bodies and manufacturers interpret the MDR differently, which is why there is still no clear consensus regarding risk class I for software.
- The official update to the MDCG guidance has done little to clarify the previous ambiguity between risk classes I and IIa. This is very good news. The bottom line is that there was at least no intention to explicitly exclude risk class I, e.g., for DiGA and exercise apps. It can be assumed that risk class I will continue to be accepted by most regulatory authorities in the future with regard to low-risk exercise apps. It remains to be seen whether the BfArM will change its opinion on this at some point.
We at QuickBird Medical are of the opinion that risk class I software products are a must. The risks posed by such products are negligible, but collectively they have a huge impact on improving patient care. A higher risk class significantly reduces the appeal of developing and approving such products. The associated costs are prohibitive for many companies or simply too risky. However, our healthcare system—and above all patients—benefit enormously from Class I risk products.
Subscribe to our newsletter to stay up to date on risk classification.
Please feel free to contact us if you are planning to develop a software medical device. We develop regulated apps, web applications, and other medical software on a contract basis for other companies.

