

Since 2019, around 73 million people with statutory health insurance in Germany have been entitled to digital health applications (DiGA): the so-called “app on prescription.” However, the interaction between DiGA and medical device regulations often causes confusion among prospective DiGA manufacturers. The requirements for medical devices and DiGA are very different, but at the same time there is a lot of overlap. It is important to understand this interface precisely.
This article clarifies important questions about the relationship between DiGA and medical devices:
- Does a DiGA have to be a medical device?
- Welche Risikoklassen sind für DiGA erlaubt?
- Which risk classes are permitted for DiGA?
- What additional requirements must DiGAs meet?
- Can a DiGA be combined with hardware medical devices?
This article is aimed at prospective DiGA manufacturers and other professionals who want to understand the regulatory requirements that apply to digital health applications. At QuickBird Medical, we have already been involved in the development of over 15 DiGAs on a contract basis and share our knowledge in specialist articles.
Table of Contents
- 1. Interaction: DiGA and Medical Devices
- 2. Which MDR Medical Device Classes are permitted for DiGA?
- 3. Medical Device vs. DiGA: Additional Requirements for DiGA
- 4. Connecting Medical Devices and Hardware to DiGA
- 5. Differences between Medical Devices and DiGA
- 6. Conclusion: Recommendations for Manufacturers
1. Interaction: DiGA and Medical Devices
The DiGA concept has created a structured path for bringing patient-oriented medical software and apps into the reimbursement system of statutory health insurance in Germany.
Prerequisite: The application must first be marketed as a medical device in accordance with the European Medical Device Regulation (MDR). Software is considered a medical device if it is intended by the manufacturer for a medical purpose, such as the diagnosis, treatment, or monitoring of diseases. Only with a valid CE mark can a manufacturer submit a DiGA application to the Federal Institute for Drugs and Medical Devices (BfArM).
In addition to being approved as a medical device, a medical application must also meet a wide range of other requirements in order to be included in the DiGA directory.

Requirements for DiGA and Software Medical Devices
1.1 So does a DiGA have to be a Medical Device?
The answer is clear: Yes, every DiGA must be a medical device.
This follows directly from the legal definition. Section 33a of the German Social Code, Book V (SGB V) defines DiGA as “low-risk medical devices whose main function is based primarily on digital technologies.” A DiGA is therefore not an alternative to a medical device, but rather a special category of medical devices with additional requirements.
1.2 What does this mean specifically for Manufacturers?
Before a manufacturer can even submit an application for inclusion in the DiGA directory to the BfArM, the conformity assessment procedure in accordance with the MDR must be fully completed. The CE marking is proof that the product meets the essential safety and performance requirements and may be placed on the market in Europe.
Important to understand: The BfArM does not check MDR compliance itself in the DiGA procedure. This is assumed and proven by the CE marking. Instead, the BfArM checks the additional requirements of the DiGAV, such as proof of positive care effects, data protection, and interoperability.
1.3 When is my Software considered a Medical dDevice?
So before the question of DiGA status becomes relevant, manufacturers must first clarify whether their software is a medical device at all. This decision is fundamental to the entire regulatory process.
Whether software is considered a medical device depends primarily on the medical purpose specified by the manufacturer. Software is only a medical device if it serves a medical purpose, i.e., if, as already mentioned, it supports the diagnosis, treatment, or monitoring of diseases.
The distinction is not always trivial. An app that simply tracks fitness data is not usually a medical device. However, the same app with a function for detecting cardiac arrhythmia is. The decisive factor is the purpose declared by the manufacturer for the product and the functions that are actually offered.
1.4 When is Software not a Medical Device?
Not every health app is automatically a medical device. The following categories, for example, are generally not covered by the MDR:
- Pure data storage or transmission: Software that only stores or transmits health data without processing or interpreting it medically.
- Administrative functions: scheduling, reminders without medical context, practice management
- General health information: Apps that provide generic information without giving individual recommendations
- Lifestyle and wellness applications: fitness trackers, nutrition apps, or meditation apps without medical purposes
Our comprehensive guide “Is your software a medical device?” addresses this topic in detail and provides specific decision-making aids.
2. Which MDR Medical Device Classes are permitted for DiGA?
Not every medical device can become a DiGA. The legislator has restricted DiGA eligibility to certain risk classes. This reflects the idea that DiGAs are designed as low-threshold care offerings with correspondingly limited risk potential.
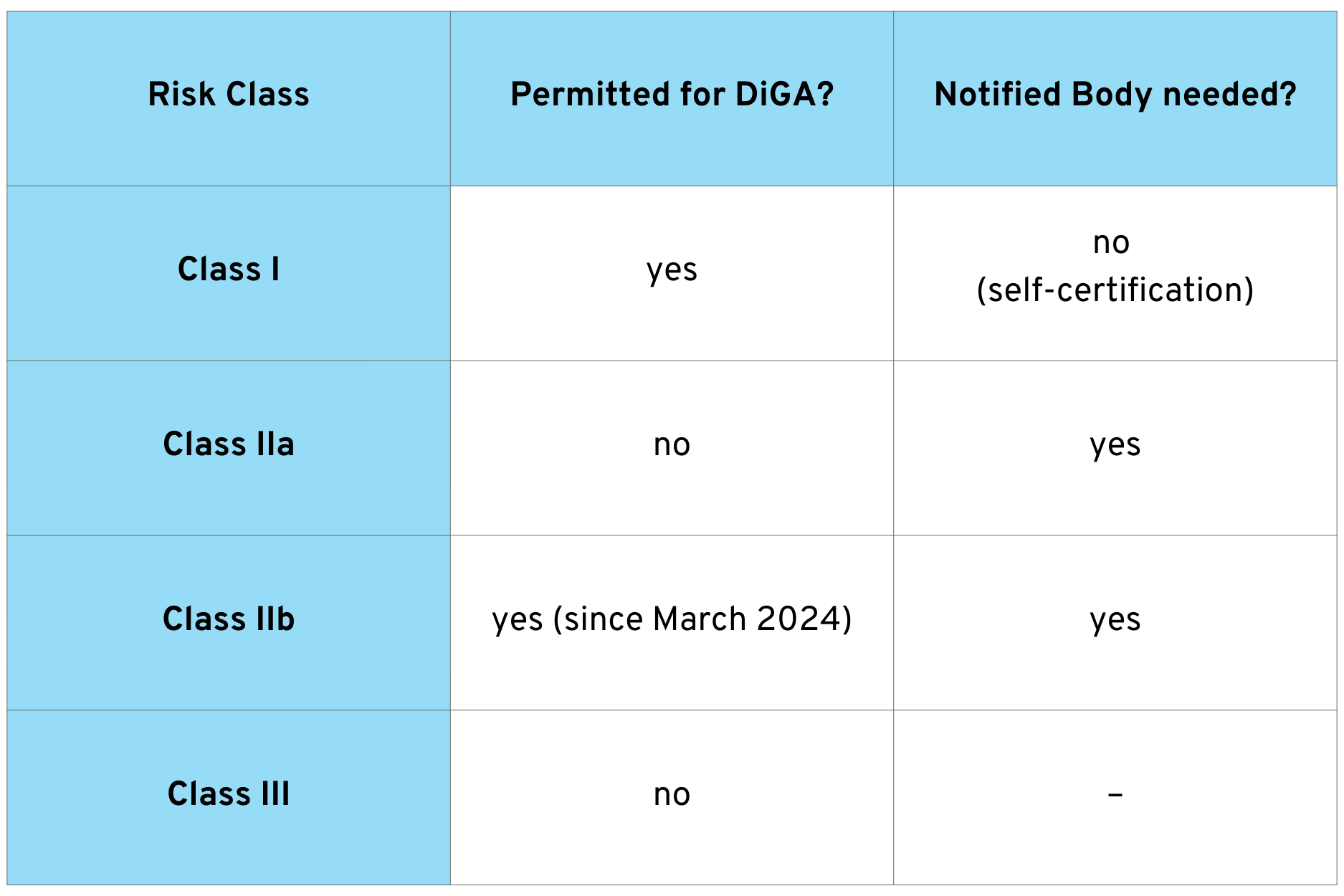
The following overview shows which risk classes are approved for DiGA:
 (Click to enlarge)
(Click to enlarge)
For Class I medical devices, the manufacturer can carry out the conformity assessment itself and issue the declaration of conformity. For Class IIa and IIb devices, however, a notified body must be involved to review the quality management system and technical documentation.
2.1 Expansion through the Digital Act (DigiG)
An important change came into effect with the Digital Act (DigiG) on March 26, 2024: Since then, medical devices in risk class IIb can also be included in the DiGA directory. Previously, only classes I and IIa were permitted.
This extension allows for DiGA with a higher risk profile. However, special requirements apply to Class IIb DiGA: for example, provisional admission for testing is not possible. Manufacturers must submit the results of a study demonstrating medical benefit when submitting their application.
2.2 How do I determine the Medical Device Risk Class for my DiGA?
Correct classification is one of the first and most important steps in developing a DiGA. It not only determines whether a notified body needs to be involved, but also influences the overall regulatory effort and costs.
Software medical devices are classified according to the rules in Annex VIII of the MDR. Rule 11 is particularly relevant for software. This rule classifies software based on, among other things, the risk posed by the information provided to the patient.
The classification depends on several factors: the type of disease (serious vs. non-serious), the type of decision support (diagnosis, therapy, monitoring), and the reversibility of possible damage caused by incorrect information.
Our article Classification of Software Medical Devices explains the classification rules in detail with practical examples.
Practical tip: Address classification at an early stage – ideally during the design phase. The risk class significantly determines the regulatory effort, costs, and time frame until market launch.
3. Medical Device vs. DiGA: Additional Requirements for DiGA
A DiGA must comply with two regulatory levels: first, the MDR requirements as a medical device and, second, the specific requirements of the DiGA Regulation (DIGAV). This section provides an overview of both levels and shows the additional hurdles that DiGA manufacturers must overcome compared to “normal” medical device manufacturers.
3.1 Requirements as a Medical Device (MDR)
Every DiGA must first meet the basic MDR requirements. These form the foundation on which the DiGA-specific requirements are based. The MDR requires a comprehensive quality and documentation system.
The key requirements include:
- Quality management system in accordance with ISO 13485, which documents and controls all quality-related processes
- Risk management according to ISO 14971, which identifies potential hazards and defines measures to minimize risk
- Software lifecycle according to IEC 62304, which structures the development and operation of software
- Usability according to IEC 62366-1, which ensures that the product can be operated safely and effectively
- Clinical evaluation demonstrating the clinical benefit and safety of the product
- Post-market surveillance, which continuously monitors the product after it has been placed on the market
These requirements apply to all medical devices. This is regardless of whether they are also to be included in reimbursement as DiGA.
Our article Approval & Certification of Software Medical Devices covers the MDR requirements in detail.
3.2 Additional Requirements for DiGA (DiGAV)
Beyond the MDR medical device requirements, the DiGAV defines specific criteria that a DiGA must meet. These requirements are reviewed by the BfArM as part of the fast-track procedure.
3.2.1 Positive Supply Effects (pVE)
DiGA must demonstrate a positive supply effect. This can be either a medical benefit (e.g., improvement in health status) or a patient-relevant structural and procedural improvement (e.g., better therapy adherence, simplified access to care).
For permanent inclusion in the DiGA directory, a comparative study is required to demonstrate the positive effect on care. In the case of provisional inclusion, the manufacturer is given a trial period of up to 12 months (up to 24 months in justified exceptional cases) to provide this evidence. However, even for inclusion on a trial basis, a comprehensive evaluation concept including a pilot study must be submitted.
More on this: Demonstrating positive effects on care provision
3.2.2 Data Protection
The DiGAV imposes particularly strict data protection requirements that go beyond those of the GDPR. Only certain, conclusively defined purposes of data processing are permitted. Special regulations apply to the processing of health data outside Germany. Transparency regarding the type and scope of data processing is mandatory.
More on this: DiGA data protection requirements
3.2.3 Data Security
An information security management system (ISMS) is mandatory for DiGA manufacturers and must even be certified according to ISO 27001.
In addition, DiGA manufacturers must obtain certification in accordance with BSI TR-03161. This is a very demanding certification for information security, which is awarded by testing bodies and finally by the BSI (Federal Office for Information Security).
More on this: BSI TR-03161 for DiGA
3.2.4 Interoperability
DiGA must use interoperable standards so that health data can be exchanged between different systems. Specifically, this means the ability to export DiGA data in human-readable and machine-readable formats (using FHIR profiles and the MIO DiGA Toolkit) and the ability to connect to electronic patient records (ePA).
More on this: Interoperability for DiGA
3.2.5 Quality Requirements
The DiGAV defines further quality requirements in various areas:
- Robustness: The DiGA must function stably and be fault-tolerant.
- Consumer protection: Transparent information, no misleading advertising, compliance with the German Medicines Advertising Act
- User-friendliness: Accessible design, intuitive operation
- Support for service providers: Meaningful integration into existing care processes
- Support for service providers: Meaningful integration into existing care processes
- Patient safety: Risk-minimizing measures, appropriate warnings
3.2.6 Application-accompanying Performance Measurement (AbEM)
After inclusion in the DiGA directory, you must continuously collect data on the use and effectiveness of your DiGA. The application-accompanying performance measurement includes usage data, patient satisfaction (PGI-C scale), and, in the future, indication-specific PROMs. This data must be reported regularly to the BfArM.
3.3 Further Information
You can find more information about the criteria you must meet to be a DiGA here: DiGA Definition & Criteria
4. Connecting Medical Devices and Hardware to DiGA
A common question from manufacturers concerns the combination of DiGA with hardware: Can a DiGA also integrate sensors, wearables, or other medical devices? The answer is nuanced. Basically yes, but under certain conditions.
DiGA are primarily software medical devices. Their main function must be based primarily on digital technologies. However, they can communicate with hardware medical devices and other systems, exchange data, and process it—as long as the main digital function predominates.
4.1 Reimbursable vs. Non-Reimbursable Hardware
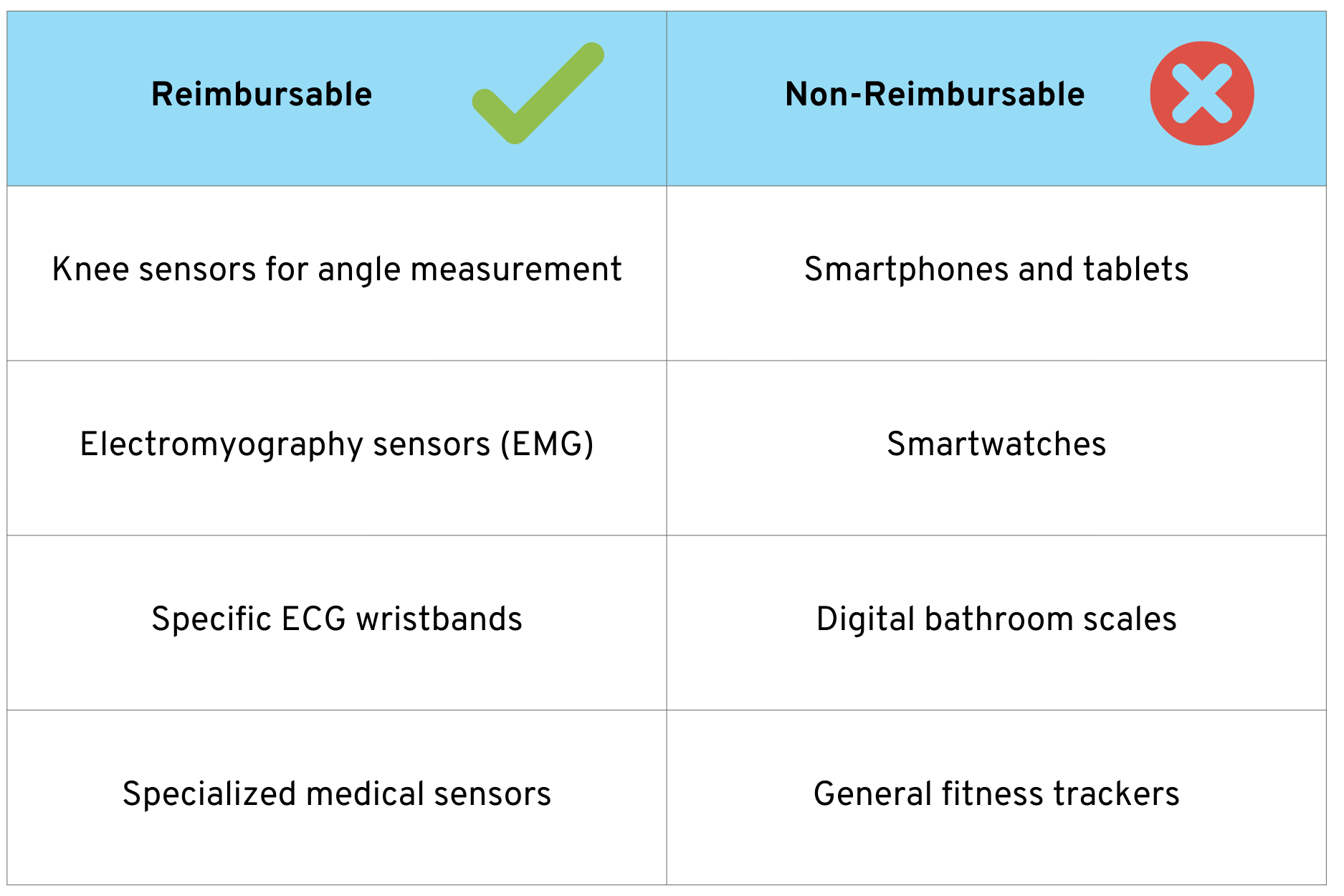
An important aspect for manufacturers is the question of reimbursement eligibility. Not all hardware that is combined with a DiGA is reimbursed by statutory health insurance.
The following overview shows examples of which types of hardware are potentially eligible for reimbursement and which are not:
 (Click to enlarge)
(Click to enlarge)
Basic rule: Specialized medical devices that are not everyday items are eligible for reimbursement. Everyday items, even if they have medical functions, are not covered by statutory health insurance, but can be used with a DiGA.
5. Differences between Medical Devices and DiGA
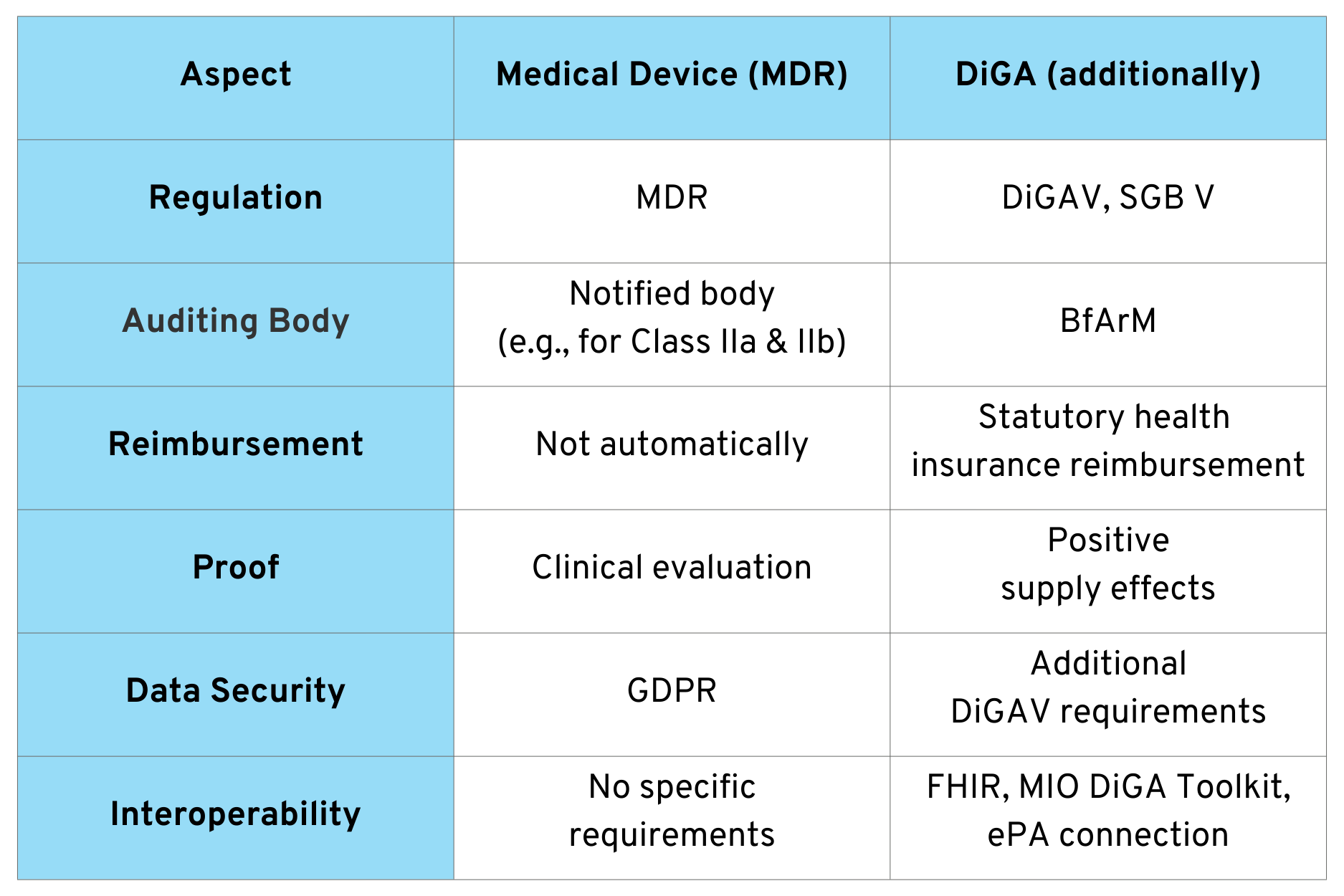
Now that we have looked at the individual aspects in detail, let’s summarize some of the key differences between a “normal” medical device and a DiGA.
 (Click to enlarge)
(Click to enlarge)
This list could be expanded considerably and is only intended to provide a basic understanding.
6. Conclusion: Recommendations for Manufacturers
The path to DiGA always leads through medical devices. Anyone who develops a digital health application and wants to have it reimbursed by statutory health insurance must consider both regulatory levels from the outset.
Our recommendations for manufacturers:
- Define your regulatory strategy early on: Clarify at the outset whether your product is a medical device and whether the DiGA route makes sense. Not every medical device has to become a DiGA.
- Ensure MDR compliance as a basis: No CE marking means no DiGA. Allow sufficient time for the conformity assessment. This applies in particular to Class IIa and IIb, where notified bodies must be involved.
- Plan DiGAV requirements in parallel: Data protection, data security, and interoperability should be incorporated into product development from the outset. Subsequent adjustments are time-consuming and expensive.
- Start planning your study for pVE verification early on: Especially if you are aiming for permanent inclusion, start designing your verification study early on.
- Take advantage of BfArM consulting: The BfArM offers paid consulting sessions. This investment is worthwhile in order to clarify any uncertainties in advance.
Are you planning a DiGA? We develop digital health applications on behalf of companies: from conception and MDR approval to inclusion in the DiGA directory. We take care of every step until successful reimbursement and beyond.

