

How does medical device approval for software work? How long does this process take? And what are the costs involved in certification?
The path to medical device approval is also complex for software products. It is therefore advisable to deal with the (self-)certification of your software early enough and to be clear about the necessary steps. After all, there is also a lot to consider apart from pure software development.
If you would like to find out what exactly you need for the approval of your software as a medical device in order to be allowed to use the CE marking, you have come to the right place.
This article discusses the approval of software medical devices under the Medical Device Regulation (MDR), which is the applicable regulation for medical devices in the EU.
Contents
- 1. Preparation of the certification
- 2. Approval of the software
- 3. Framework conditions for the approval of software medical devices
- 4. Conclusion
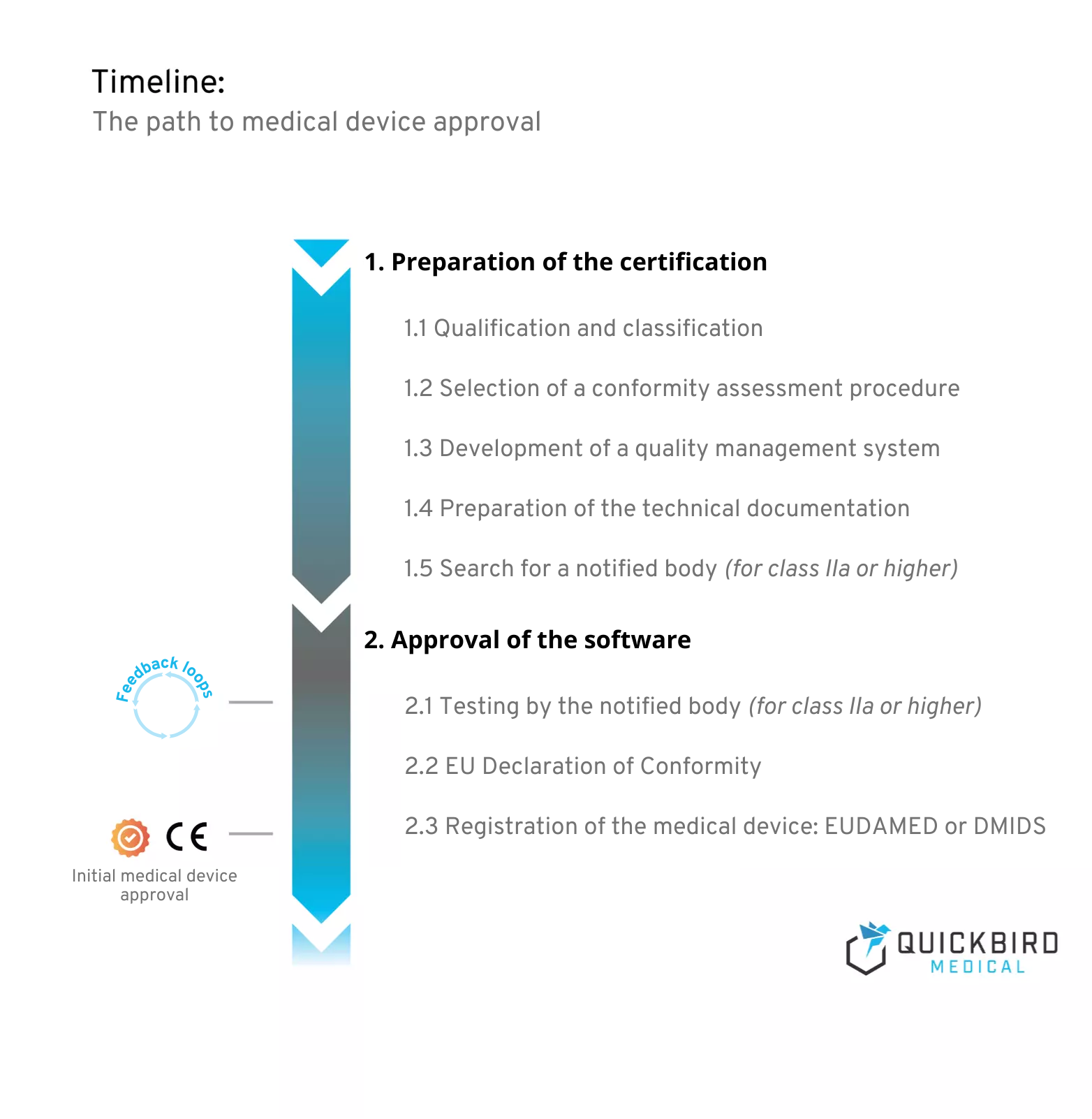
Schedule for medical device approval
1. Preparation of the certification
Preparation for certification always begins before the actual development of your medical device software. This is because the effort required for smooth approval depends heavily on the risk class that you define in the first steps of the project.
The latter decides on the conformity assessment procedure to be applied and the need for a notified body for approval. What is necessary in all cases, however, is the establishment of a quality management system. This is intended to ensure the quality of your product and is the basis for the technical documentation that must be prepared for medical devices of all risk classes.
1.1 Qualification and classification
At the very beginning of any medical software is the definition of the intended use – more precisely, the “intended purpose”. This is because it fundamentally determines whether the planned product is a medical device at all (qualification). The next step is to classify the product accordingly and assign it to a risk class (I, IIa, IIb or III). This article focuses on the approval and certification of your software medical device.
A detailed excursus into the qualification and classification of software would go beyond the scope of this article. Of course, we have written our own articles on these topics, which you can find here:
- Phrasing of the intended purpose for (software) medical devices
- Guide: Is your app a medical device?
- Classification of software medical devices: MDR Guideline
It is important that you carry out the classification of your software conscientiously and deal with this topic at an early stage. This is because the risk class determines, among other things, the conformity assessment procedure to be used, which is discussed later in this article and has a significant impact on the effort, duration and costs of the project.
If you have not yet established a quality management system in your company, the next step is to set one up (regardless of the risk class). This article explains what you need to bear in mind when developing medical devices: Guidelines for the development of medical apps: What manufacturers need to pay attention to
1.2 Selection of a conformity assessment procedure
‘conformity assessment’ means the process demonstrating whether the requirements of this Regulation relating to a device have been fulfilled;
(Quote from article 2 MDR)
In order for your software to bear a CE marking, manufacturers must first carry out a so-called conformity assessment procedure. The MDR offers three different assessment procedures from Annex IX to XI. The procedure to be used must also be selected during the planning phase of the project.
At this point, we will say straight away that the only practicable procedure for software is actually the one in Annex IX: conformity assessment on the basis of a quality management system and an assessment of the technical documentation.
Although it is also possible to carry out an assessment procedure on the basis of a so-called “type examination”, this does not make sense in most cases. In a document (NB-MED/2.2/Rec4), which states the position of the notified bodies on this topic, it says verbatim:
“[…] a pure product related evaluation without consideration of the design process is not considered adequate. Consequently, the use of some of the Conformity Assessment Procedures (CAP), as defined by the directives may be unsuitable for software.”
The document also describes the need for life cycle processes. This would basically mean that product development must follow the specifications of IEC 62304, which also requires a quality management system. The question therefore arises as to why manufacturers do not choose the conformity assessment procedure in accordance with Annex IX.
The application of the conformity assessment procedure in Annex IX (and the involvement of a notified body) is not required for manufacturers of class I medical devices. However, this does not mean that you do not have to do anything for a class I product – you still need a quality management system and the technical documentation for your product. However, this is not checked by a notified body.
But what kind of software still falls into class I? Read more about this in this article: Classification of software medical devices: MDR Guidance
In addition to the conformity assessment procedure, as a manufacturer you must also sign an EU declaration of conformity for your product.
Info: Difference between quality management system and technical documentation:
- A quality management system describes the process landscape in your company and is product-independent. Processes are workflows that are initiated and run through by certain triggers and always generate one or more outcomes. Risk management would be a classic example of a process (which can of course also be subdivided into several sub-processes).
- Technical documentation, on the other hand, is product-specific and contains all documents and records relating to the product. This includes, for example, plans and reports (e.g. for risk management or clinical evaluation).
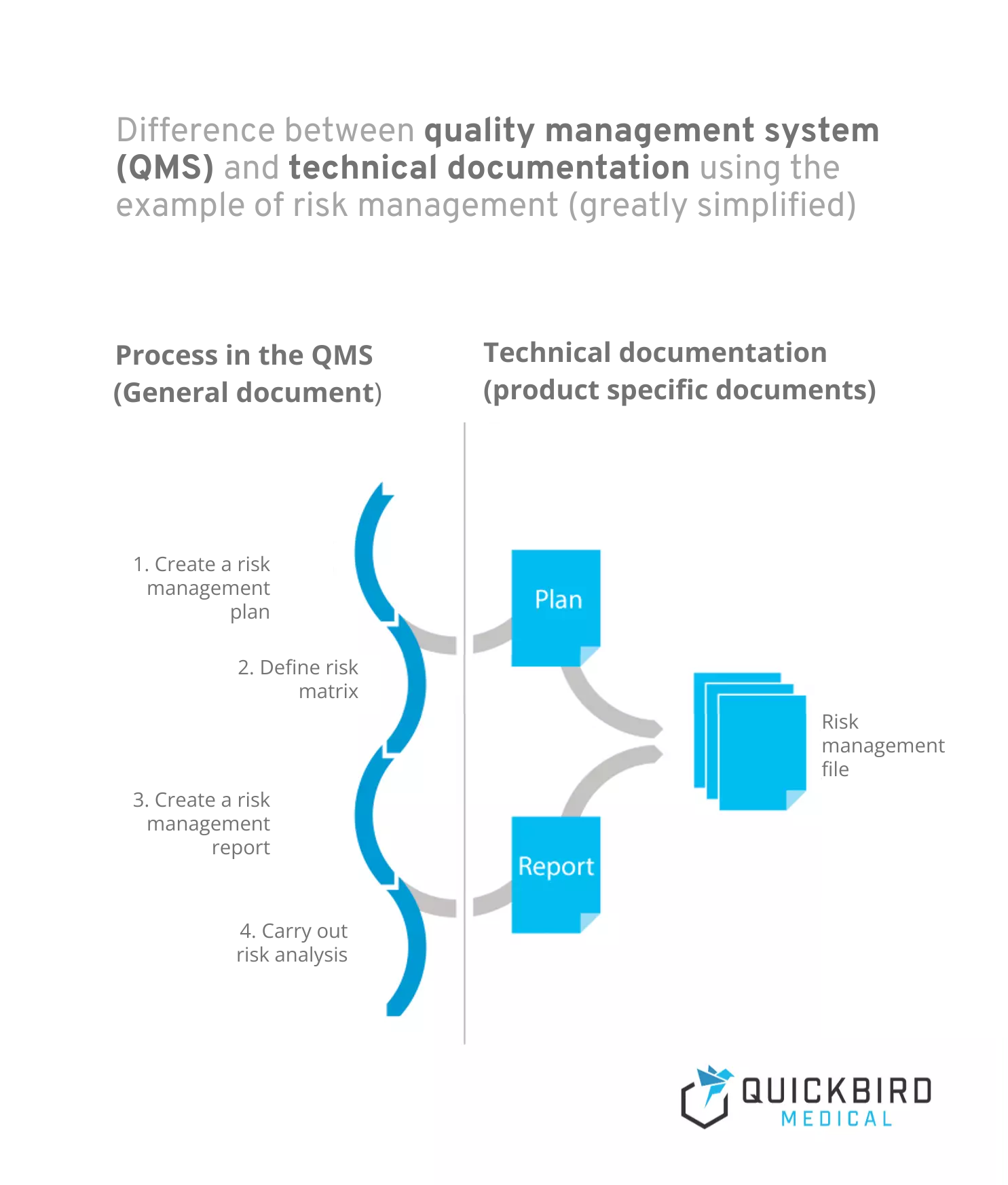
Difference between quality management system and technical documentation
1.3 Development of a quality management system
Is your planned product a medical device? Congratulations, you will soon have your own quality management system! This is mandatory for all software medical device manufacturers and forms the basis for the majority of the technical documentation that you must create for your product in accordance with the MDR.
While the MDR is not really a good guide for setting up a QMS, it is best to start with ISO 13485 – this is also not a good guide, but at least it is more specific in many places with regard to the requirements. However, you will not receive a step-by-step guide that tells you exactly what you need to do. Therefore, you will most likely need external support in the form of consulting services. Experience has shown that it takes several months to set up a QMS, depending on the size of the company and the complexity of existing structures.
Please note: Overall, the requirements of the MDR for quality management systems are somewhat more extensive than those of ISO 13485. However, the standard provides a good basis and covers most of the requirements of the MDR.
In this article, you can find out which other standards you should take into account when developing medical software products: Guidelines for the development of medical apps: What manufacturers need to pay attention to.
1.4 Creation of technical documentation
The technical documentation is specified in Annexes II and III of the MDR. It is primarily intended to help you as a manufacturer to develop better products in terms of performance and patient safety and includes, for example, planning documents, reports and records that are created during the product life cycle.
The description of planned activities after placing on the market must also be taken into account here. Much of the technical documentation comes from the processes of your quality management system, which means that much of it is generated automatically if you stick to your processes. Roughly speaking, technical documentation is a collection of all information relating to the product..
1.4.1 Conduct of a clinical trial
In many cases, you will also need to conduct a clinical trial for your product. This is also part of the technical documentation and falls within the scope of clinical evaluation. Read more about clinical evaluation in this article: MDR-Guide: Clinical evaluation of software medical devices.
1.5 Search for a notified body (for class IIa or higher)

When a notified body is necessary
Info: There may also be rare exceptional cases in which a notified body is also required for class I medical devices (e.g. reusable surgical instruments). However, this does not apply to software in the vast majority of cases.
Products of class IIa or higher require the “go” of a notified body before they can be placed on the market with a CE marking. And this is where the planning of medical device certification can hit a major snag. A delayed search for a notified body can delay the approval of your software by several months – a scenario that you naturally want to avoid as a medical device manufacturer. There are 2 main reasons why we recommend that you start your search early:
- Demand is greater than supply: Waiting times of several months are quite common. Specifically, there are currently only 8 notified bodies in Germany that can carry out an inspection in accordance with the MDR (as of October 2022). All notified bodies according to MDR can be found in the Nando directory or on OpenRegulatory.
- Not all notified bodies have an equal affinity for software. In other words, some departments make the process unnecessarily difficult for you because they don’t understand that software development differs from classic medical device production in many ways. It saves you a lot of time, and above all nerves, if you get an auditor who is familiar with software products and “speaks your language”. In addition, software-savvy departments also have a better understanding of which measures make sense in a QMS and there is less chance that they will focus on processes that are of little relevance to software during the audit.
The search for a suitable named entity therefore usually begins in the Nando directory linked above. We do not wish to make a recommendation for or against any of the notified bodies at this point, but the following sources may be helpful when selecting a notified body:
- If possible, get testimonials from other software medical product manufacturers
- Ask explicitly about experience with software medical devices in initial consultations with the notified bodies
- You already have an ISO 13485 certificate? Check whether your certification body is also certified as a notified body (assuming you are satisfied with it)
- Get in touch with us
If you are also aiming for ISO 13485 certification, it makes sense to apply for this from an organization that is also certified as a notified body – this way you get both from a single source, do not have to keep changing your certification bodies and do not fall victim to different interpretations by the individual organizations.
2. Approval of the software
Have you established a quality management system and created your technical documentation? Great! In this chapter, you will learn what you need to pay attention to before you can launch your product on the market.
2.1 Testing by the notified body (for class IIa or higher)
As described above, a notified body must be involved in the approval of your medical device for products in class IIa or higher. Two things are audited (usually in this order):
- The technical documentation
- Your quality management system
The review of the technical documentation can include several feedback loops and, depending on requirements, can extend over several weeks and months. As soon as the technical documentation has been approved, your quality management system will be audited (this sequence is usual, but not mandatory).
As a rule of thumb, the larger your company, the longer the on-site audit by the notified body will take. You should therefore be prepared for several days of visits by the auditors (in exceptional cases, the audit may also be carried out remotely).
You hold an ISO 13485 certificate?
In purely formal terms, ISO 13485 certification makes no difference – it neither shortens the audit of your quality management system by the notified body nor eliminates it altogether.
This is because the scope of ISO 13485 is not entirely congruent with that of the MDR when it comes to quality management systems. You can also obtain ISO certification from organizations that are not certified as notified bodies. Nevertheless, an ISO 13485 certificate is of course an advantage because your auditor will approach you with a completely different feeling if you have one.
This signals that your quality management system must be “already in order” on the whole and that the probability of major deviations is lower. In addition, you will have less trouble explaining why you believe your quality management system is MDR-compliant.
Tip: If you do not yet have an ISO 13485 certificate for your quality management system, it is advisable to apply for one as part of the QMS audit. Ask your designated body whether this is possible. This may save you a second audit. Legally, however, you do not need an ISO 13485 certificate.
2.1.1 Special case for risk class I
As mentioned above, manufacturers of class I products do not need a notified body to assess the conformity of their software. In this case, it is sufficient to establish a quality management system, prepare the technical documentation in accordance with Annex II and III and then draw up the EU Declaration of Conformity.
The software can then be registered as a medical device in DMIDS or EUDAMED (more on this below) and you have fulfilled your obligations as a manufacturer. This means that the product can be placed on the market with a clear conscience with the CE marking.
But is there no audit by the supervisory authority?
Not necessarily – at least market entry is not dependent on it. Officially, the authorities are not obliged to check such products in detail when they are registered. However, when registering a product for the first time, it is quite realistic that the technical documentation and/or your QMS will at least be randomly checked.
You should therefore be prepared to be asked by your regulatory authority to provide access to your technical documentation (e.g. risk management or clinical evaluation) some time after the product has been notified. However, you do not have to wait for such a request – you can already place your product on the market.
You can find out how to determine the risk class of your medical device software in this article: Classification of software medical devices: MDR guide
2.2 Your EU Declaration of Conformity
Before entering the market, you as the manufacturer declare the conformity of your product in writing. This document contains all the information defined in Annex IV of the MDR. You can download a corresponding template for the EU Declaration of Conformity here.
EU Declaration of Conformity – free download
We will send you a template for the EU Declaration of Conformity free of charge via e-mail. Simply enter your e-mail address.
2.3 Registration of the medical device: EUDAMED and DMIDS
Do I have to register my product with the BfArM (DMIDS) or in the EUDAMED database? Or even in both systems? This question is currently facing all medical device manufacturers, as it is planned that EUDAMED will replace DMIDS in the future. At the moment, the answer is pretty quick: DMIDS is compulsory, EUDAMED is voluntary.
Note: This is the status as of 03/25/2022. Changes in the meantime could not be taken into account.
3. Framework conditions for the approval of software medical devices
3.1 How long does the approval process take?
The time required for the certification of your medical device cannot, of course, be determined on a flat-rate basis. Nevertheless, there are a few important parameters that you need to take into account when planning. The time frame given in brackets should be regarded as a rule of thumb – in reality, this information may also vary:
- Development of a quality management system (several months)
- Product development & creation of technical documentation (several months to years)
- Clinical trial, if necessary (several months to years)
- For class IIa or higher:
- Search for a designated position (up to 6 months)
- Inspection of the technical documentation and the QMS by the notified body (approx. 3-12 months)However, this does not mean that you do not have to do anything for a class I product – you still have to set up a QMS and create the technical documentation. However, this is not checked by a notified body.
You have probably noticed that the need for a designated body is one of the most important time factors. This means that the approval of class I medical devices takes comparatively little time.
Tip: If you need a notified body, the following measures can help to minimize the time required:
- Make an appointment with a notified body as early as possible to check the technical documentation for your product. Of course, it is not possible to estimate to the day when your technical documentation will be final. But it is better to make a planned appointment and postpone it if necessary than not to have one at all. However, do not forget to inform your designated body immediately if you are unable to meet the deadline.
- Choose a notified body that has a certain affinity for software medical devices. This reduces communication difficulties and prevents impractical adjustments to your QMS and other documents.
- Make sure you submit technical documentation that is as error-free as possible. In particular, you should correct clerical errors and obvious inconsistencies yourself in advance. This reduces the number of feedback loops, which usually last several days or weeks.
Overall, you should plan several months for the review of the technical documentation and the auditing of the quality management system.
3.2 How much does the approval cost?
The costs for medical device certification can vary greatly, so we list here the cost items that usually carry the most weight. Basically, it can also be said here that a class I product is the cheapest option. This is because from class IIa you need a notified body for the conformity assessment of your software, which naturally increases the costs. Generally speaking, it is worth mentioning the costs for
- Consulting services for setting up a quality management system
- UDI numbers for products (usually a few 100€/year)
- Clinical trial, if necessary (several 10,000€ or more)
- (only for Class IIa or higher:) the notified body (€50,000 – €200,000). Here it depends very much on how “satisfied” the notified body is with your technical documentation and the quality management system. The more effort (e.g. feedback loops) is required on the part of the notified body, the higher the costs. The size of the company and the number of products are also a decisive factor for the costs.
When we talk about costs, we must of course also consider the time factor in addition to the financial outlay. On the one hand, setting up a quality management system takes at least a few months in any case. As a result, you will probably lose out on sales in the short term – even if you must of course see a compliant quality management system as a sensible investment in the future. After all, this is the foundation of every software medical device manufacturer.
Another time-consuming factor affects manufacturers of Class IIa or higher products: a notified body always blocks the market entry of medical devices for several months. This means that you cannot earn anything from the product until certification has been completed by the notified body. You can find out exactly what you can expect from a risk class IIa product compared to class I in a separate technical article we have published: MDR risk class I vs. IIa: Differences for software medical devices
So consider in advance whether these costs can realistically be borne by your company. Start-ups in particular often underestimate the effort involved and frequently do not have the necessary resources to survive this “revenue-free” period.
3.3 What do I need to consider for products with artificial intelligence (AI)?
If you want to integrate artificial intelligence (AI) into your product, there are a few other aspects to consider when approving the medical device. In our comprehensive guide on this topic, we explain in detail what is particularly relevant for you when certifying AI products: To the AI guide
4. Conclusion
The most important key message is this: The effort, duration and costs for the approval of your medical device software stand and fall with the risk class. For all classes, however, a quality management system must be set up and the technical documentation for the product must be prepared.
The big difference is that, as a manufacturer, you will be scrutinized much more intensively from risk class IIa onwards. Manufacturers of class I medical devices are, with a lot of luck, only inspected by their supervisory authority many months after entering the market (and then often only superficially), whereas other manufacturers have to be certified by a notified body.
This checks both the technical documentation of your product and your quality management system. You should expect the costs for the notified body to be around €30,000-35,000 (depending on the effort involved, these costs can of course be much higher) – but don’t forget the major internal effort required to set up a quality management system! You will certainly have to invest several months and a lot of working time here.
Do not underestimate these costs and be aware that unforeseen delays of several months may occur in the course of medical device approval before you generate your first sales with your product.
The following articles are also relevant for you if you are planning to develop a medical software product:
- Phrasing of the intended purpose for (software) medical devices
- Guide: Is your app a medical device?
- Classification of software medical devices: MDR Guideline
- Artificial intelligence (AI) in medical devices – MDR guidelines (2024)
You can find even more articles in our blog.

