

Anyone involved in the development of software medical devices is certainly no stranger to the term “Quality management system” (QMS). You may also have heard of ISO 13485 in this context.
ISO 13485 is a standard that defines the requirements for a quality management system for medical device manufacturers. The core objective is to pursue the goal of developing safe and functional products.
But how exactly can this be achieved? What is a process-based approach? What are the contents of ISO 13485 and how do I implement them in my software company?
In this article, we answer the most important questions about the content, requirements and chapters of ISO 13485 and about quality management systems in general.
Overview
- 1. Quality management system – explained simply
- 2. ISO 13485 – Guidance for software manufacturers
- 3. Frequently asked questions (FAQ)
- 4. Other relevant standards for software manufacturers
- 5. Medical device approval without a quality management system?
- 6. Conclusion
1. Quality Management System – explained simply
Before you start implementing ISO 13485, let’s take a brief look at what a quality management system actually is. A quality management system describes the entirety of all processes in your company that are necessary to implement products in accordance with product requirements.
The term quality here basically describes how well the result (product) matches the product requirements (e.g. patient safety, medical benefit, customer satisfaction).
Let’s start with a very simplified example that makes the meaning of a quality management system (according to ISO 13485) more tangible:
You want to produce a scalpel and you purchase the metal (raw material) from a supplier. You are looking for steel online and find a good offer on ebay from an anonymous supplier, which you buy immediately. Using your grinding stone from the kitchen, you shape the lump and the medical product is completed.
The scalpel would now be sharp and ready for use. Would you undergo open heart surgery with it? Hopefully not.
In compliance with ISO 13485, the production of the scalpel would not be possible in such an uncontrolled manner. In this simplified example, you would have to document the following processes and, of course, comply with them:
- “Determination of requirements related to product” – First of all, you need to identify what specific requirements are placed on your scalpel. These can be, for example, requirements from (potential) customers or legal regulations. (Chapter 7.2.1 of ISO 13485)
- “Purchasing process” – Evaluation of the supplier providing the metal based on relevant criteria. A private supplier on ebay would probably not pass this (chapter 7.4.1 of ISO 13485)
- “Verification of purchased product” – You must check that the raw material you are purchasing precisely meets your requirements (verification) (chapter 7.4.3 of ISO 13485)
- “Work environment and contamination control” – Ensuring a suitable working environment. A private kitchen has numerous uncontrollable influences that could jeopardize the quality of the product (chapter 6.4 of ISO 13485)
- “Infrastructure” – Ensure suitable working equipment. Your old grindstone is probably not suitable for scalpels (chapter 6.3 of ISO 13485)
- “Human resources” – Ensuring the competence of personnel. As an accountant, you may not have the necessary qualifications to process steel (Chapter 6.2 of ISO 13485)
- … and so on (chapters 4 to 8 of ISO 13485)
As you can see, ISO 13485 is strict and controls pretty much everything that could have an impact on the quality of the product. Companies that comply with ISO 13485 therefore inevitably endeavor to leave nothing to chance in order to guarantee the consistent quality of their products – simply what the name “quality management” states.
However, as ISO 13485 is a very general standard and applies equally to all medical device manufacturers, it is often written in a vague, cryptic and perhaps even incomprehensible way – especially for software manufacturers.
For example, what is a production process for an app? Do I now have to deal with contamination in our office? And how am I supposed to prove that my software is delivered in a sterile condition???
Questions upon questions – this blog article is intended to shed light on the subject and make it easier for software manufacturers to implement ISO 13485.
2. ISO 13485 – Guidance for software manufacturers
As described at the beginning, ISO 13485 is a standard for all manufacturers of medical devices. Regardless of whether we are talking about scalpels, prostheses or software products.
That is why we would like to provide better orientation in this guide and highlight the key points for software manufacturers in each chapter.
2.1 Process-based approach
ISO 13485 follows a process-based approach. As the name suggests, this means that you have to write and apply processes.
These processes cover the entire life cycle of a medical device – from planning through to decommissioning and also include areas that are only indirectly related to the product.
A process requires inputs and generates outputs, which in turn are or can be inputs for other processes
An example of a process could be risk management, which requires the inputs shown in the figure (e.g. medical purpose) and generates certain outputs (e.g. risk management plan):
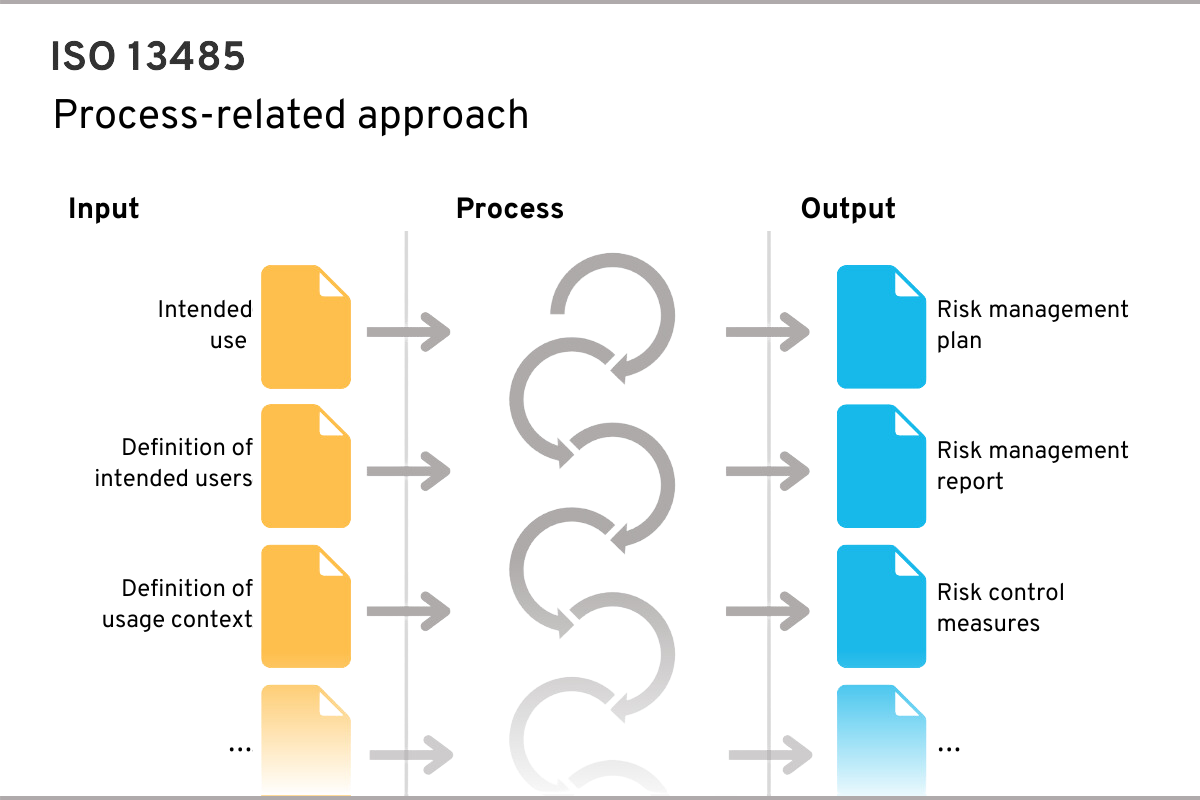
The process-based approach of ISO 13485
2.2 First steps – Starting from scratch
When you open an ISO standard for the first time, you might immediately think about developing a classic lifestyle app to avoid the strict medical device regulations.
But even if ISO 13485 seems overwhelming at first – it’s not that bad after all. Once you have understood how to read the standard, you will find it much easier to implement it.
Note: We strongly recommend consulting an experienced advisor when setting up a quality management system.
Not only can they help you interpret the standard, they can also work with you to draw up a project plan and support you in implementing the standard specifically in your company with all its particularities.
2.3 How to read ISO 13485?
ISO 13485 is actually very easy to handle – if you know how. You can basically regard each sub-chapter from the main chapters 4, 5, 6, 7 and 8 as a separate requirement that requires explicit implementation. The table of contents is therefore the ideal basis for appropriate mapping.
Now you only have to go through the theory once from top to bottom and think about how to implement each point.
Especially if you have no previous experience, we do NOT recommend writing the entire quality management system from scratch. It is much more expedient to spend some money and buy appropriate templates. You can find these online, although we do not recommend any provider at this point..
Of course, purchased templates are usually very generic unless they have been created specifically for your company by a consulting firm.
You will therefore have to make adjustments in some areas so that the processes can also be applied to your company. However, customizing templates is usually less error-prone and leads to better results than the starting-from-scratch approach.
A consultant can also help you customize templates so that you can achieve a good result for your company through several review loops.
Make absolutely sure that you actually fulfill all the criteria before you put the quality management system into practice and start developing your medical device software.
Now enough of the introductory words – let’s take a closer look at the contents of ISO 13485.
2.4 Contents of ISO 13485
ISO 13485 comprises a total of 8 chapters, whereby no specific implementation measures can be derived from the first 3. Here you will find the scope of ISO 13485, references to other relevant standards and the definitions.
That is why in this guide we are focusing on those chapters that have practical implications for you as a manufacturer. As described above, you will find all the requirements to be implemented in chapters 4, 5, 6, 7 and 8.
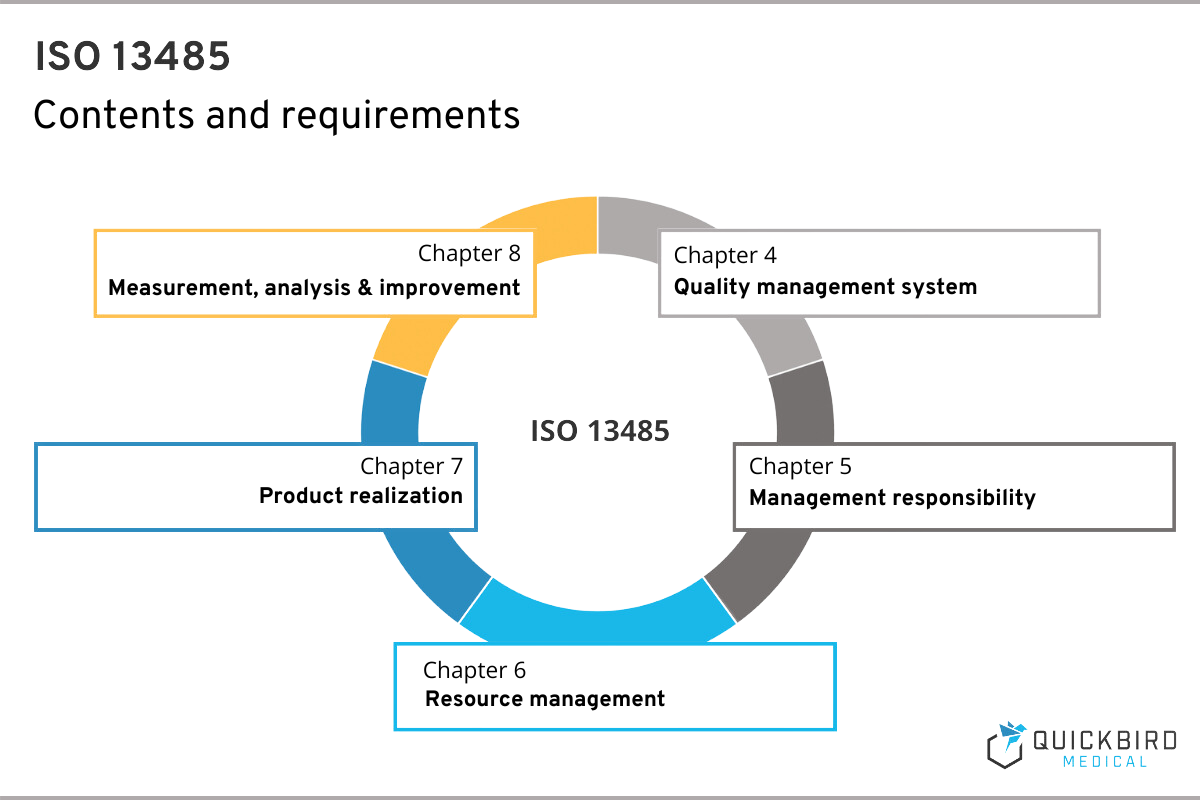
Contents & requirements of ISO 13485
Important: It is possible to declare individual requirements from chapters 6, 7 and 8 as “not applicable” if these are not applicable to your company. In the software environment, this includes, for example, the special requirements for sterile medical devices (7.5.5). However, if you do not apply certain chapters, you must give a reason for it.
2.4.1 Chapter 4 – Quality Management System
In this chapter you will find “general requirements” for your quality management system, as well as “documentation requirements”.
Chapter 4, for example, requires the identification and implementation of all necessary processes to be implemented by your company. Most of these can be found in ISO 13485 itself, but there are also processes from other standards and regulations that are relevant here (e.g. MDR, IEC 62304 or ISO 14971).
To do this, it is necessary to understand the role(s) of your company in the market in order to be able to define the area of application of the quality management system.
You will also find general requirements for your processes and the quality management system in this chapter. The focus here is on the task of assessing the Effectiveness of the quality management system. This is done, for example, by providing the necessary resources or assigning certain responsibilities and roles.
A central feature of the quality management system is the obligation to document pretty much everything. You will also find corresponding requirements for the control (e.g. creation, release, ensuring availability, etc.) of documents and records in chapter 4.
Another requirement of Chapter 4 is the creation of a quality management manual and a medical device file. The former provides an overview of the quality management system, so to speak. The scope of application is clearly defined there and the process landscape is presented.
The quality management manual has no clear product reference, but refers to the quality management system itself.
The medical device file, on the other hand, contains (or refers to) the documentation that demonstrates the conformity of individual types or groups of medical devices.. Contents include, for example, the intended purpose and a list of product requirements.
As you can see, Chapter 4 primarily sets out the basic rules on which the entire quality management system is based.
2.4.2 Chapter 5 – Management responsibility
This chapter is primarily about one thing: Commitment.
For example, the management is obliged to create the necessary framework conditions for an effective quality management system and to identify the applicable regulatory and customer requirements.
This is to be achieved by defining quality objectives, a quality policy and the appointment of a “management representative”. In addition, the management must ensure appropriate communication channels within the company (e.g. Slack, e-mail, etc.).
Note: “Management” usually refers to senior management.
The obligation to carry out management reviews (management assessments) is also key. These take place at a defined interval (e.g. annually) and aim to ensure the ongoing suitability, appropriateness and effectiveness of the quality management system.
This assessment cannot be carried out by feel, which is why this management assessment must also be carried out on the basis of data from various sources (e.g. customer feedback). The management assessment should therefore result in measures that improve quality management and the products themselves. In addition, the need for further resources in the company is to be determined.
2.4.3 Chapter 6 – Resource Management
Staff, skills, laptops, Git, … these are just a few examples of resources you may need to develop a software medical device. Since software companies rarely have supply chains or factory floors, the focus is primarily on human and digital resources, which is why Chapter 6 is a strong HR topic, among others.
Here, particular emphasis is placed on the qualification of employees, which must be ensured by your company. This has an impact on the selection of new employees as well as on the training and development of existing staff.
And even if modern software companies manage without a fixed workplace and their employees are only given a laptop as their only necessary work tool, the issue of “infrastructure” should not be neglected.
This infrastructure is then digital, but still central to the development of safe and functional medical device software. You must determine and document the requirements for this infrastructure.
Software manufacturers, on the other hand, do not usually have to concern themselves with the working environment and contamination. However, it is important to document a corresponding (short) statement for the “exclusion” of this requirement. This is required both by the standard itself and by some auditors.
2.4.4 Chapter 7 – Product Realization
Chapter 7 is where it gets exciting, because this is the part that you will be dealing with most intensively if you want to establish a quality management system in accordance with ISO 13485.
This is about the actual product that is to be developed – from planning to completion. The first step is to identify customer requirements and ensure that these can be implemented.
This also includes requirements that are not explicitly set by your customers (e.g. regulatory requirements).
Chapter 7 describes how to plan product realization, identify product requirements and finally evaluate, verify and validate the results.
At this point, reference should be made to IEC 62304, which manufacturers of software medical devices should always comply with. This standard precisely describes the procedure for software development and, in addition to ISO 13485, places additional requirements on software development.
Product development according to ISO 13485 proceeds roughly as follows:
- Development planning
- Development inputs
- Development results
- Development assessment
- Development verification
- Development validation
- Transfer of development to production
- Control of development changes
The order of the chapters suggests that these activities follow each other in strict sequence. However, this is not necessarily the case in reality. Some activities can run in parallel (e.g. development planning and determination of product requirements (development inputs)).
Furthermore, there is no requirement that all these activities be performed only once. This may make sense in the hardware sector, but in agile software development it is perfectly possible to run some of these activities in a cycle.
IEC 62304 must also be complied with when implementing software products – read more about this in this article (IEC 62304: Software life cycle processes for medical devices).
Note: What confuses many manufacturers is the distinction between “development verification” and “development validation“
After the actual development, product realization also includes the verification and validation of the product. As this distinction is often unclear, here is a small rule of thumb to help you understand the difference. The difference is best illustrated by two questions:
- Verification: Has the product been implemented correctly?
- Validation: Has the right product been implemented?
During the verification process, the result is compared with the clear product requirements – for example, “The app shows the user therapy recommendations” (e.g. through software system tests)
During the validation, it is checked whether the result can also fulfill its purpose – for example, “The therapy recommendations alleviate the disease” (e.g. through a clinical study).
Find out more about validation in this article: Validation of medical device software according to MDR
You will also find requirements for the identifiability and traceability of products in chapter 7, which is best ensured using a versioning system. If monitoring and measuring equipment is used (e.g. special testing tools), these must also be handled in accordance with ISO 13485.
Procurement
In addition to the actual development of the product, Chapter 7 also deals with the regulation of purchased products and services. This involves evaluating suppliers and verifying the services purchased.
For example, if your development team works with Jira, you must first evaluate Atlassian (its manufacturer) as a supplier and then investigate whether Jira fulfills your requirements for a ticket management system. You may only use Jira in your company after successful evaluation and validation. You can find out more about the validation of software used in our article: Validation of software according to MDR
2.4.5 Chapter 8 – Measurement, Analysis, and Improvement
As soon as your new quality management system is up and running, you will soon see that the processes are not yet working smoothly in some places and may even lead to regulatory deviations.
The handling of optimizations and corrections is dealt with in chapter 8 – because of course you also need your own processes for this. However, it is not only data about the quality management system itself that needs to be collected and processed, but also, of course, data about your medical devices in particular.
In this chapter you will therefore find criteria for data analysis, monitoring, dealing with non-conforming products and continuous improvements to your quality management system.
In addition to regular monitoring audits, you are also obliged to carry out internal audits to ensure the ongoing conformity of your quality management system.
2.5 Update 2021 with amendment: ISO 13485:2016/A11:2021
In 2021, ISO 13485 was updated by amendment A11:2021. ISO 13485/A11:2021 did not change the main part of ISO 13485. The main new addition is an official mapping to the MDR. This can be found in Annex ZA.
This mapping is very helpful in analyzing and identifying open requirements and should therefore be taken into account by manufacturers.
In summary, this means that amendment A11:2021 does not impose any new or amended requirements on the quality management system, but primarily supports the achievement of MDR compliance.
Since the new version is harmonized, you should now use EN ISO 13485:2016/A11:2021 or DIN EN ISO 13485:2021 as the basis for your MDR products.
2.6 QMS software, Atlassian, and others – The right tooling
As you can see, a quality management system consists of numerous documents and records that are interrelated. A very networked system, which can be made clearer with the appropriate tooling.
For many people, a quality management system still sounds like a lot of paper. Of course, you can work with printed documents and approve them with a signature, for example.
Generally, however, this is not the method of choice – software companies in particular, whose employees do not always work in a central location, encounter problems with this approach.
Of course, there are now also numerous digital options for implementing a quality management system. A few options for implementation are listed in the table below.
Advantages
| Method | Disadvantages | Comments | |
| Printed documents (e.g. Microsoft Word) |
|
|
This method is particularly common in older companies. In software companies in particular, however, it is not the method of choice in the vast majority of cases. |
| Special software for QMS (e.g. Greenlight Guru) |
|
|
Special software can be useful in some cases – especially if the costs are not a major factor at first. |
| Cloud-based document management software (e.g. Confluence/Jira) |
|
|
This method is very flexible and generally much cheaper than special software for QMS. It is therefore particularly interesting for start-ups. |
| Text editor with digital version management software (e.g. Word + Git) |
|
|
This method is also inexpensive and easy to customize. However, it requires a greater technical understanding than the other methods. |
3. Frequently asked questions (FAQ)
3.1 Who needs ISO 13485?
ISO 13485 is used by medical device manufacturers and contains requirements for a quality management system. This is required, for example, by the MDR in order to be able to develop medical devices.
3.2 What does an ISO 13485 certificate confirm?
An ISO 13485 certificate confirms that a company possesses a quality management system for medical devices. This is required by law if you wish to manufacture and distribute medical devices in the EU (and in many other countries). However, it does not serve as proof that you comply with all MDR requirements.
3.3 Is ISO 13485 mandatory?
No, ISO 13485 is merely a recognized standard for quality management systems for medical devices. However, anyone wishing to develop medical devices under the MDR is well advised to obtain the relevant certification. In particular, a notified body will scrutinize your quality management system and without certification it could be difficult for you to argue your case. An ISO 13485 certificate is also helpful for manufacturer monitoring by supervisory authorities.
3.4 What do I need to bear in mind with products featuring artificial intelligence (AI)?
If you want to integrate artificial intelligence (AI) into your product, there are a few additional aspects to consider when implementing ISO 13485. In our comprehensive guide on this topic, we explain in detail what is particularly relevant for you when certifying AI products: To the AI guide
3.5 What is the relationship between ISO 13485 and the MDR?
According to the MDR, all medical device manufacturers must have a quality management system. However, the MDR does not specify what exactly this should look like. This is why ISO 13485 has become the industry standard in Europe when it comes to quality management system requirements in the medical device sector. Of course, ISO 13485 is not a 1:1 implementation guide for the MDR, because there are also requirements that arise directly from the MDR and are not directly covered by ISO 13485 (for example, the appointment of a “Person responsible for regulatory compliance – PRRC”, or a process for reporting incidents to competent authorities).
Important: At no point does the MDR refer to the implementation of ISO 13485, but always to a “quality management system”. There is therefore no formal obligation to comply with ISO 13485. However, appropriate certification is strong proof of the conformity of your quality management system, which is why we highly recommend it. Most MDR auditors also expect this.
3.6 Can the manufacture of medical devices be outsourced?
Yes, if you want to focus on sales and marketing, for example, you can also outsource the manufacture of the medical device to a service provider – such as QuickBird Medical. This means that the service provider assumes the role of the distributor and legally places the medical device on the market. Regulatory responsibility is therefore transferred to the service provider..
3.7 Download ISO 13485 as a PDF?
ISO 13485 can be purchased inexpensively at https://www.evs.ee/en and can be downloaded as a PDF (in English). Of course, you can also obtain the standard on numerous other platforms, but usually at higher prices. You can find the German version of ISO 13485 at Beuth: https://www.beuth.de/de/norm/din-en-iso-13485/332674603
4. Other relevant standards for software manufacturers
In addition to ISO 13485, as a manufacturer of software medical devices you must also deal with and implement other standards. IEC 62304, which places requirements on software lifecycle processes, should be mentioned here first and foremost. You can find out more in this article: IEC 62304: Software life cycle processes for medical devices
. It is also worth taking a look at IEC 82304, especially for manufacturers of standalone software. Although there are numerous references to the aforementioned IEC 62304, it contains additional requirements for the validation of healthcare software.
You will also notice that in some places reference is made to risk management as an accompanying process. The best way to implement a corresponding process is in accordance with ISO 14971, which is supplemented by IEC 62366 (application of usability engineering for use to medical devices). You can find more information in this article: Guideline for the development of medical apps: What manufacturers need to look out for
Of course, there are other standards that could be relevant for you. Our list only includes those that we believe require the greatest implementation effort.
5. Medical device approval without a quality management system?
Is there a way to completely avoid regulatory obligations for medical device manufacturers?
Yes, there is: outsourcing the role of legal manufacturer to an external company. This is an efficient and risk-minimizing solution that conserves your resources while ensuring regulatory compliance. You don’t need to set up a quality management system and can leave the regulatory approval process entirely to us.
This allows you to focus on product design and marketing, while QuickBird Medical, as the legal manufacturer, takes care of all regulatory requirements and is responsible for compliance.
You can find more information here: To the technical article
6. Conclusion
ISO 13485 is probably the most widely used standard for setting up quality management systems for medical device manufacturers in Europe. If you comply with these, you cover most of the requirements of the MDR with regard to quality management systems.
The sub-chapters in chapters 4 to 8 each describe individual requirements that you as a manufacturer must implement and therefore already provide the ideal basis for a corresponding roadmap.
Even though ISO 13485 is written in a way that is easy to understand, its implementation is not entirely self-explanatory. The specifics of individual companies must always be taken into account, which is why there is no one-size-fits-all quality management system that all software companies can apply equally.
Nevertheless, it is advisable to buy appropriate templates and then adapt them.
We also recommend that you set up your quality management system together with an experienced consultant. Appropriate software tools can also give you a significant efficiency advantage.
Nevertheless, setting up a quality management system is very time-consuming and generates costs. It may therefore make sense for you to (initially) hand over the role of distributor for your product to a service provider. They then assume regulatory responsibility for your product and are legally responsible for its conformity. In this case, you avoid the obligation to set up your own quality management system.
If you are planning to implement a medical software product and/or set up a quality management system, please do not hesitate to contact us. We support you in your project. Contact us now.

