

Wer sich mit der Entwicklung von Software-Medizinprodukten beschäftigt, dem ist der Begriff “Qualitätsmanagementsystem” (QMS) bestimmt nicht fremd. Vielleicht haben Sie in diesem Zusammenhang auch schon von der ISO 13485 gehört.
Die ISO 13485 ist eine Norm, welche Anforderungen an ein Qualitätsmanagementsystem für Medizinprodukt-Hersteller definiert. Im Kern dabei soll das Ziel verfolgt werden, sichere und funktionstaugliche Produkte zu entwickeln.
Doch wie genau soll das gelingen? Was ist ein prozessbezogener Ansatz? Was sind die Inhalte der ISO 13485 und wie setze ich sie in meinem Software-Unternehmen um?
In diesem Artikel beantworten wir die wichtigsten Fragen zu den Inhalten, Anforderungen und Kapiteln der ISO 13485 und zu Qualitätsmanagementsystemen im Allgemeinen.
Überblick
- 1. Qualitätsmanagementsystem – einfach erklärt
- 2. ISO 13485 – Leitfaden für Software-Hersteller
- 3. Frequently asked questions (FAQ)
- 4. Weitere relevante Normen für Software-Hersteller
- 5. Medizinprodukt-Zulassung ohne Qualitätsmanagement-System?
- 6. Fazit
1. Qualitätsmanagementsystem – einfach erklärt
Bevor Sie sich an die Umsetzung der ISO 13485 setzen, sehen wir uns vorab kurz an, was ein Qualitätsmanagementsystem eigentlich ist. Ein Qualitätsmanagementsystem beschreibt die Gesamtheit aller Prozesse in Ihrem Unternehmen, die dazu notwendig sind, um Produkte gemäß den Produktanforderungen umzusetzen.
Der Begriff Qualität beschreibt hier im Grunde, wie gut das Ergebnis (Produkt) mit den Produktanforderungen (z.B. Patientensicherheit, medizinischer Nutzen, Kundenzufriedenheit) übereinstimmt.
Lassen Sie uns mit einem stark vereinfachten Beispiel beginnen, welches den Sinn eines Qualitätsmanagementsystems (nach ISO 13485) greifbarer macht:
Sie möchten ein Skalpell herstellen, wobei Sie das Metall (Rohstoff) von einem Lieferanten beziehen. Sie suchen online nach Stahl und finden ein gutes Angebot auf ebay von einem anonymen Anbieter, bei dem Sie sofort zuschlagen. Mit Ihrem Schleifstein aus der Küche bringen sie den Klumpen in Form und fertig ist das Medizinprodukt.
Das Skalpell wäre nun scharf und einsatzbereit. Würden Sie sich damit am offenen Herzen operieren lassen? Hoffentlich nicht.
Unter Einhaltung der ISO 13485 wäre die Herstellung des Skalpells nicht so unkontrolliert möglich. Sie müssten nämlich in diesem abgespeckten Beispiel schonmal auf jeden Fall die folgenden Prozesse dokumentieren und natürlich auch einhalten:
- Ermittlung der Produktanforderungen – Zunächst müssen Sie identifizieren, welche konkreten Anforderungen an Ihr Skalpell gestellt werden. Das können z.B. Forderungen von (potenziellen) Kunden, oder auch gesetzliche Vorschriften sein. (Kapitel 7.2.1 der ISO 13485)
- Bewertung des Lieferanten, welcher das Metall liefert anhand relevanter Kriterien – ein Privatanbieter auf ebay würde dieser vermutlich nicht standhalten (Kapitel 7.4.1 der ISO 13485)
- Bewertung des eingekauften Rohstoffs – Sie müssen überprüfen, dass der Rohstoff, den Sie einkaufen, auch genau Ihren Anforderungen entspricht (Verifizierung) (Kapitel 7.4.3 der ISO 13485)
- Sicherstellung einer geeigneten Arbeitsumgebung – eine private Küche birgt zahlreiche unkontrollierbare Einflüsse, welche die Qualität des Produkts gefährden könnten (Kapitel 6.4 der ISO 13485)
- Sicherstellung geeigneter Arbeitsausrüstung – Ihr alter Schleifstein ist für Skalpelle vermutlich nicht geeignet (Kapitel 6.3 der ISO 13485)
- Sicherstellung der Kompetenz des Personals – Als Buchhalter bringen Sie ggf. nicht die notwendigen Qualifikationen mit, um Stahl zu bearbeiten (Kapitel 6.2 der ISO 13485)
- … und so weiter (Kapitel 4 bis 8 der ISO 13485)
Man sieht also, die ISO 13485 ist streng und kontrolliert so ziemlich alles, was einen Einfluss auf die Qualität des Produkts haben könnte. Unternehmen, die die ISO 13485 einhalten, sind also zwangsläufig darum bemüht, möglichst nichts dem Zufall zu überlassen, um die konstante Qualität Ihrer Produkte zu gewährleisten – eben einfach das, was auch schon der Name “Qualitätsmanagement” sagt.
Da die ISO 13485 aber eine sehr allgemeine Norm ist und für alle Medizinprodukthersteller gleichermaßen gilt, ist sie an vielen Stellen schwammig, kryptisch und vielleicht sogar unverständlich geschrieben – besonders für Software-Hersteller.
Was soll ein zum Beispiel ein Produktionsprozess für eine App sein? Muss ich mich in unserem Büro jetzt mit Kontamination beschäftigen? Und wie soll ich nachweisen, dass meine Software in sterilem Zustand ausgeliefert wird???
Fragen über Fragen – dieser Blogartikel soll Licht ins Dunkel bringen und die Umsetzung der ISO 13485 für Software-Hersteller erleichtern.
2. ISO 13485 – Leitfaden für Software-Hersteller
Wie eingangs beschrieben, ist die ISO 13485 eine Norm für alle Hersteller von Medizinprodukten. Ganz gleich, ob es um Skalpelle, Prothesen oder eben Softwareprodukte geht.
Deshalb möchten wir in diesem Leitfaden eine bessere Orientierung geben und pro Kapitel aufzeigen, was die zentralen Punkte für Software-Hersteller sind.
2.1 Prozessbezogener Ansatz
Die ISO 13485 verfolgt einen prozessbezogenen Ansatz. Wie der Name schon sagt, bedeutet das für Sie: Sie müssen Prozesse schreiben und anwenden.
Diese Prozesse erstrecken sich über den gesamten Lebenszyklus eines Medizinprodukts – Begonnen bei der Planung, bis hin zur Außerbetriebnahme und umfassen auch Bereiche, die nur indirekt mit dem Produkt zu tun haben.
Ein Prozess benötigt jeweils Eingaben und erzeugt Ausgaben, welche wiederum die Eingaben für andere Prozesse sind oder sein können.
Ein Beispiel für einen Prozess könnte Risikomanagement sein, welcher die in der Abbildung dargestellten Eingaben (z.B. Medizinische Zweckbestimmung) benötigt und bestimmte Ausgaben (z.B. Riskomanagement-Plan) erzeugt:
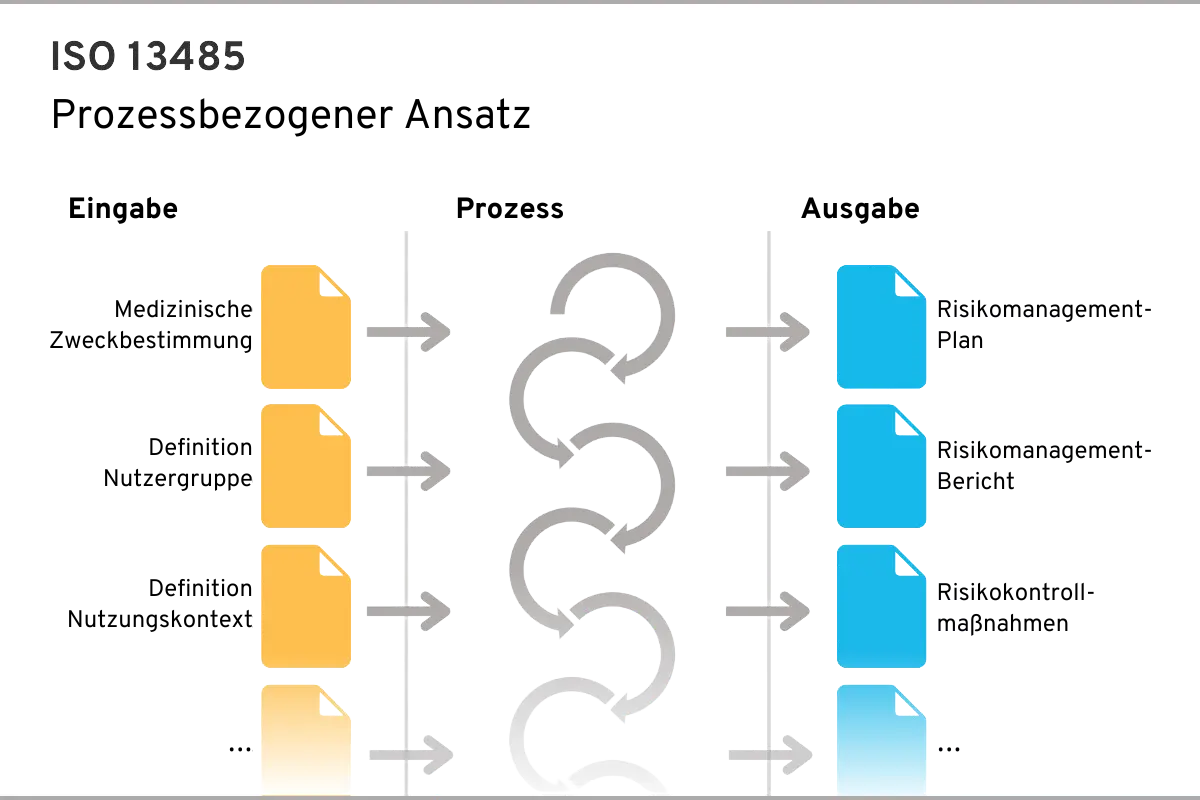
Der prozessbasierte Ansatz der ISO 13485
2.2 Erste Schritte – Starting from scratch
Wenn Sie zum ersten Mal eine ISO Norm aufschlagen, werden Sie vielleicht schlagartig darüber nachdenken, doch lieber eine klassische Lifestyle-App zu entwickeln, um den strengen Medizinprodukt-Vorschriften zu entgehen.
Doch auch wenn die ISO 13485 im ersten Moment überwältigend wirkt – so schlimm ist sie dann doch nicht. Sobald Sie verstanden haben, wie die Norm zu lesen ist, werden Sie es weitaus leichter haben, diese auch umzusetzen.
Hinweis: Wir empfehlen stark, für den Aufbau eines Qualitätsmanagementsystems einen erfahrenen Berater hinzuzuziehen.
Dieser kann nicht nur bei der Interpretation der Norm helfen, sondern erstellt mit Ihnen gemeinsam einen Projektplan und unterstützt Sie auch bei der Umsetzung der Norm speziell in Ihrem Unternehmen mit all seinen Besonderheiten.
2.3 Wie liest man die ISO 13485?
Die ISO 13485 ist an sich sehr angenehm zu handhaben – wenn man weiß, wie. Sie können jedes Unterkapitel aus den Hauptkapiteln 4, 5, 6, 7 und 8 im Grunde als eigene Anforderung betrachten, welche eine explizite Umsetzung erfordert. Das Inhaltsverzeichnis stellt daher schon die optimale Basis für ein entsprechendes Mapping dar.
Nun müssen Sie in der Theorie nur einmal von oben bis unten durchgehen und sich bei jedem Punkt überlegen, wie Sie diesen umsetzen.
Vor allem, wenn Sie keine Vorerfahrungen haben, empfehlen wir NICHT, das gesamte Qualitätsmanagementsystem von Grund auf neu zu schreiben. Viel zielführender ist es, etwas Geld in die Hand zu nehmen und sich entsprechende Templates zu kaufen. Diese finden Sie online, wobei wir an dieser Stelle keinen Anbieter empfehlen.
Natürlich sind eingekaufte Templates meist sehr generisch, sofern sie nicht eigens für Ihr Unternehmen durch eine Consulting-Firma erstellt wurden.
Daher werden Sie an einigen Stellen Anpassungen vornehmen müssen, damit die Prozesse auch auf Ihr Unternehmen anwendbar sind. Das Anpassen von Templates ist aber meist weniger fehleranfällig und führt zu besseren Ergebnissen, als der Starting-from-scratch Ansatz.
Bei der Anpassung von Templates kann Ihnen ebenfalls ein Berater helfen, damit Sie durch mehrere Review-Schleifen zu einem guten Ergebnis für Ihr Unternehmen kommen.
Stellen Sie unbedingt sicher, dass Sie auch wirklich alle Kriterien erfüllen, bevor Sie das Qualitätsmanagementsystem in die Praxis bringen und mit der Entwicklung Ihrer Medizinprodukt-Software beginnen.
Nun aber genug der einleitenden Worte – sehen wir uns die Inhalte der ISO 13485 genauer an.
2.4 Inhalte der ISO 13485
Die ISO 13485 umfasst insgesamt 8 Kapitel, wobei sich aus den ersten 3 keine konkreten Umsetzungsmaßnahmen ableiten lassen. Hier finden Sie den Anwendungsbereich der ISO 13485, Verweise auf andere relevante Normen sowie die Begriffsbestimmungen.
Deshalb fokussieren wir uns in diesem Leitfaden auf jene Kapitel, welche praktische Implikation für Sie als Hersteller beinhalten. Wie oben beschrieben, finden Sie alle umzusetzenden Anforderungen in den Kapiteln 4, 5, 6, 7 und 8.
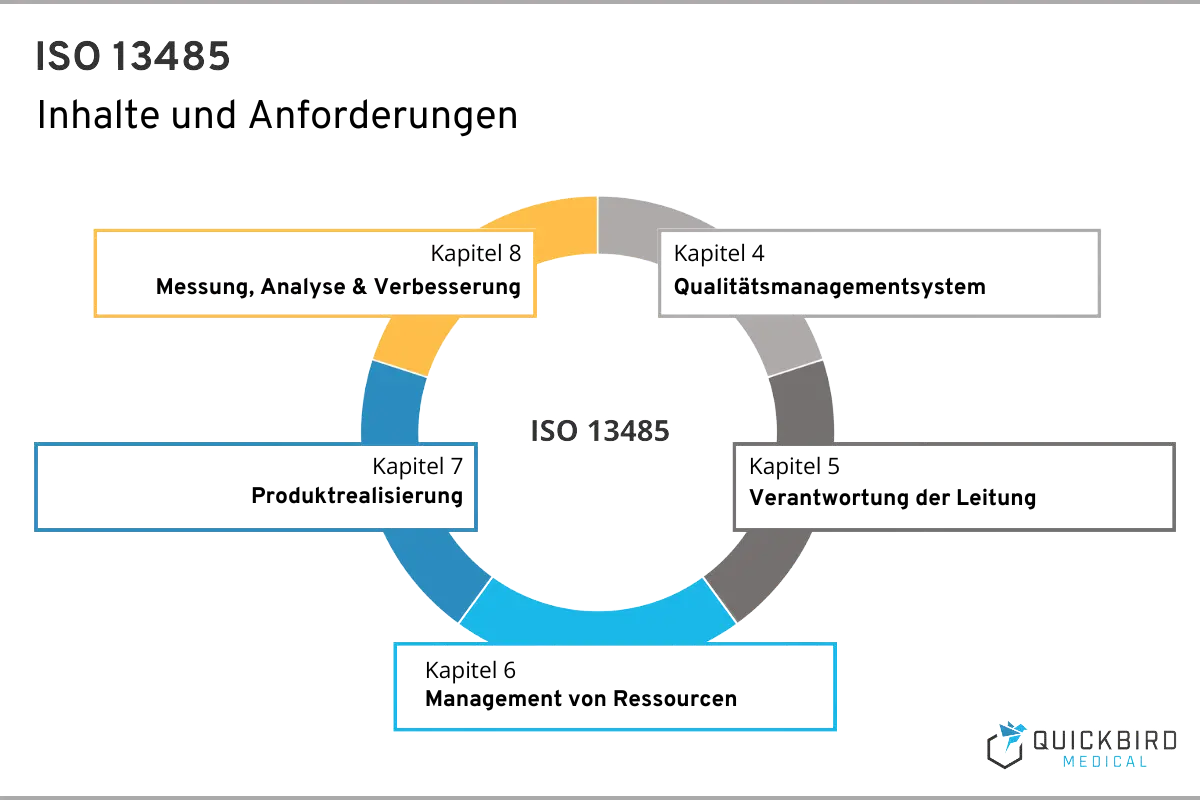
Inhalte & Anforderungen der ISO 13485
Wichtig: Es ist möglich, einzelne Anforderungen aus den Kapiteln 6, 7 und 8 als “nicht anwendbar” zu deklarieren, wenn diese für Ihr Unternehmen nicht einzuhalten ist. Im Software-Umfeld umfasst dies zum Beispiel die besondere Anforderungen an sterile Medizinprodukte (7.5.5). Wenn Sie bestimmte Kapitel aber nicht anwenden, müssen Sie eine Begründung dafür anführen.
2.4.1 Kapitel 4 – Qualitätsmanagementsystem
In diesem Kapitel finden Sie “Allgemeinen Anforderungen” an Ihr Qualitätsmanagementsystem, sowie “Dokumentationsanforderungen”.
Kapitel 4 erfordert etwa die Identifikation und Umsetzung aller notwendigen Prozesse, die von Ihrem Unternehmen umzusetzen sind. Die meisten davon finden Sie in der ISO 13485 selbst, jedoch gibt es auch Prozesse aus anderen Normen und Regularien, die hier von Bedeutung sind (z.B. MDR, IEC 62304 oder ISO 14971).
Dazu ist es notwendig, die Rolle(n) Ihres Unternehmens im Markt zu verstehen, um das Anwendungsgebiet des Qualitätsmanagementsystems definieren zu können.
Zudem finden Sie in diesem Kapitel generelle Anforderungen an Ihre Prozesse und das Qualitätsmanagementsystem. Im Fokus hierbei steht die Aufgabe, die Wirksamkeit des Qualitätsmanagementsystems sicherzustellen. Dies geschieht etwa durch die Bereitstellung erforderlicher Ressourcen oder die Vergabe bestimmter Verantwortlichkeiten und Rollen.
Eine zentrale Eigenschaft des Qualitätsmanagementsystems ist die Pflicht, so ziemlich alles zu dokumentieren. Auch für die Lenkung (z.B. Erstellung, Freigabe, Sicherstellung der Verfügbarkeit, etc.) von Dokumenten und Aufzeichnungen finden Sie in Kapitel 4 entsprechende Anforderungen.
Eine weitere Forderung von Kapitel 4 ist die Erstellung eines Qualitätsmanagement-Handbuchs und einer Medizinproduktakte. Ersteres stellt sozusagen einen Überblick über das Qualitätsmanagementsystems dar. Dort wird der Anwendungsbereich klar definiert und die Prozesslandschaft dargestellt.
Das Qualitätsmanagement-Handbuch hat keinen klaren Produktbezug, sondern bezieht sich auf das Qualitätsmanagementsystem selbst.
Die Medizinproduktakte hingegen enthält (oder verweist auf) jene Dokumentation, welche die Konformität einzelner Medizinprodukttypen oder auch -gruppen darlegt. Inhalte sind beispielsweise die Zweckbestimmung sowie eine Liste von Produktanforderungen.
Sie sehen also, Kapitel 4 stellt in erster Linie grundlegende Regeln dar, auch welchen das gesamte Qualitätsmanagement aufbaut.
2.4.2 Kapitel 5 – Verantwortung der Leitung
In diesem Kapitel geht es vor allem um eines: Commitment.
Beispielsweise wird die Leitung dazu verpflichtet, die notwendigen Rahmenbedingungen für ein effektives Qualitätsmanagementsystem zu schaffen und die anwendbaren regulatorischen- und Kundenanforderungen zu identifizieren.
Dies soll etwa durch die Definition von Qualitätszielen, einer Qualitätspolitik und der Ernennung eines “Beauftragten der Leitung” erfolgen. Außerdem muss die Leitung für angemessene Kommunikationskanäle im Unternehmen sorgen (z.B. Slack, E-Mail, etc.).
Hinweis: “Leitung” bezieht sich meist auf das obere Management .
Zentral ist zudem die Pflicht zur Durchführung von Management Reviews (Managementbewertungen). Diese finden in einem definierten Intervall statt (z.B. jährlich) und haben das Ziel, die fortdauernde Eignung, Angemessenheit und Wirksamkeit des Qualitätsmanagementsystems sicherzustellen.
Diese Bewertung lässt sich nicht nach Gefühl vornehmen, deshalb muss diese Managementbewertung auch auf der Basis von Daten aus verschiedenen Quellen durchgeführt werden (z.B. Kundenfeedback). Aus der Managementbewertung sollen demnach Maßnahmen herauskommen, welche das Qualitätsmanagement und die Produkte selbst verbessern. Zudem soll der Bedarf an weiteren Ressourcen im Unternehmen ermittelt werden.
2.4.3 Kapitel 6 – Management von Ressourcen
MitarbeiterInnen, Fähigkeiten, Laptops, Git, … das alles sind nur einige Beispiele von Ressourcen, die Sie möglicherweise benötigen, um ein Software-Medizinprodukt zu entwickeln. Da es in Software-Unternehmen selten Lieferketten oder Fabrikhallen gibt, stehen vor allem die personellen und digitalen Ressourcen im Fokus, weshalb Kapitel 6 unter anderem ein starkes HR-Thema ist.
Hier wird besonders die Qualifikation von MitarbeiterInnen betont, welche durch Ihr Unternehmen sichergestellt werden muss. Das hat einerseits Auswirkungen auf die Selektion neuer MitarbeiterInnen, als auch auf das Training und die Weiterbildung bestehenden Personals.
Und auch wenn moderne Software-Unternehmen ohne festen Arbeitsplatz auskommen und deren MitarbeiterInnen lediglich einen Laptop als einzig notwendiges Arbeitsgerät bekommen, ist das Thema “Infrastruktur” nicht zu vernachlässigen.
Diese ist dann eben digital, aber dennoch zentral für die Entwicklung sicherer und funktionsfähiger Medizinprodukt-Software. Sie müssen Anforderungen an diese Infrastruktur ermitteln und dokumentieren.
Mit der Arbeitsumgebung und Kontamination hingegen müssen sich Software-Hersteller in der Regel nicht beschäftigen. Wichtig ist es aber, ein entsprechendes (kurzes) Statement für den “Ausschluss” dieser Anforderung zu dokumentieren. Dies wird einerseits von der Norm selbst, als auch von einigen Auditoren gefordert.
2.4.4 Kapitel 7 – Produktrealisierung
In Kapitel 7 wird es spannend, denn das ist der Teil, mit dem Sie sich am intensivsten beschäftigen werden, wenn Sie ein Qualitätsmanagementsystem nach ISO 13485 etablieren möchten.
Hier geht es nämlich um das eigentliche Produkt, das entwickelt werden soll – von der Planung bis zur Fertigstellung. Zunächst geht es darum, die Kundenanforderungen zu identifizieren und sicherzustellen, dass diese auch umgesetzt werden können.
Dazu zählen auch Anforderungen, die nicht explizit von Ihren Kunden gestellt werden (z.B. regulatorische Anforderungen).
In Kapitel 7 wird beschrieben, wie Sie die Produktrealisierung planen, die Produktanforderungen identifizieren und die Ergebnisse am Ende auch bewerten, verifizieren und validieren.
An dieser Stelle sei auf die IEC 62304 verwiesen, welche von Herstellern von Software-Medizinprodukten auf jeden Fall eingehalten werden sollte. Diese Norm beschreibt das Vorgehen bei der Software-Entwicklung präzise und stellt ergänzend zur ISO 13485 noch einmal zusätzliche Anforderungen an die Software-Entwicklung.
Die Produktentwicklung läuft nach ISO 13485 grob wie folgt ab:
- Entwicklungsplanung
- Entwicklungseingaben
- Entwicklungsergebnisse
- Entwicklungsbewertung
- Entwicklungsverifizierung
- Entwicklungsvalidierung
- Übertragung der Entwicklung an die Herstellung
- Lenkung von Entwicklungsänderungen
Die Reihenfolge der Kapitel suggeriert, dass diese Tätigkeiten streng aufeinander folgen. Das ist in der Realität aber nicht zwangsläufig der Fall. Einige Tätigkeiten können durchaus parallel ablaufen (z.B. Entwicklungsplanung und Ermittlung der Produktanforderungen (Entwicklungseingaben)).
Außerdem gibt es keine Forderung, dass all diese Tätigkeiten nur ein einziges mal durchlaufen werden. Im Hardware-Bereich macht das möglicherweise Sinn, aber in der agilen Software-Entwicklung ist es durchaus möglich, einige dieser Tätigkeiten in einem Zyklus laufen.
Bei der Realisierung von Software-Produkten ist zudem die IEC 62304 einzuhalten – Lesen Sie mehr dazu in diesem Artikel (IEC 62304: Software-Lebenszyklus-Prozesse von Medizinprodukten).
Hinweis: Was viele Hersteller verwirrt, ist die Unterscheidung zwischen “Entwicklungsverifizierung” und “Entwicklungsvalidierung”
Die Produktrealisierung umfasst nach der eigentlichen Entwicklung auch die Verifizierung und Validierung des Produkts. Da diese Unterscheidung oft unklar ist, hier eine kleine Faustregel, um den Unterschied besser zu verstehen. Der Unterschied lässt sich am besten anhand zweier Fragen festmachen:
- Verifizierung: Wurde das Produkt richtig umgesetzt?
- Validierung: Wurde das richtige Produkt umgesetzt?
Bei der Verifizierung gleicht man das Ergebnis mit den klaren Produktanforderungen ab – zum Beispiel “Die App zeigt dem Nutzer Therapieempfehlungen” (z.B. durch Software-System-Tests)
Bei der Validierung wird überprüft, ob das Ergebnis seinen Zweck auch erfüllen kann – zum Beispiel “Die Therapieempfehlungen lindern die Erkrankung” (z.B. durch eine klinische Studie).
Mehr zur Validierung erfahren Sie in diesem Artikel: Validierung von Medizinprodukt-Software nach MDR
Zudem finden Sie in Kapitel 7 Anforderungen an die Identifizierbarkeit und Rückverfolgbarkeit von Produkten, was Sie am besten über ein Versionierungssystem gewährleisten. Falls Überwachungs- und Messmittel verwendet werden (z.B. spezielle Testing Tools), sind auch diese gemäß ISO 13485 zu behandeln.
Beschaffung
Neben der eigentlichen Entwicklung des Produkts geht es in Kapitel 7 auch um die Regulierung eingekaufter Produkte und Dienstleistungen. Dabei geht es um die Bewertung von Lieferanten und die Verifizierung der eingekauften Leistungen.
Wenn Ihr Entwicklerteam beispielsweise mit Jira arbeitet, müssen Sie einerseits Atlassian (dessen Hersteller) als Lieferanten bewerten und anschließend untersuchen, ob Jira die von Ihnen gestellten Anforderungen an ein Ticket-Managementsystem erfüllt. Erst nach erfolgreicher Bewertung und Validierung dürfen Sie Jira in Ihrem Unternehmen einsetzen. Mehr zur Validierung von eingesetzter Software erfahren Sie in unserem Artikel: Validierung von Software nach MDR
2.4.5 Kapitel 8 – Messung, Analyse und Verbesserung
Sobald Ihr neues Qualitätsmanagementsystem im Einsatz ist, werden Sie bald sehen, dass die Prozesse an einigen Stellen noch nicht reibungslos funktionieren und vielleicht sogar zu regulatorischen Abweichungen führen.
Der Umgang mit Optimierungen und Korrekturen wird in Kapitel 8 behandelt – denn natürlich brauchen Sie auch hierfür eigene Prozesse. Doch nicht nur Daten über das Qualitätsmanagementsystem selbst, sondern natürlich insbesondere zu Ihren Medizinprodukten müssen entsprechend gesammelt und verarbeitet werden.
In diesem Kapitel finden Sie daher Kriterien für die Datenanalyse, Überwachung, Umgang mit nicht-konformen Produkten, sowie stetige Verbesserungen Ihres Qualitätsmanagementsystems.
Neben den regelmäßigen Überwachungsaudits sind Sie außerdem dazu verpflichtet, interne Audits durchzuführen, um die fortlaufende Konformität Ihres Qualitätsmanagementsystems sicherzustellen.
2.5 Update 2021 mit Amendment: ISO 13485:2016/A11:2021
2021 wurde die ISO 13485 durch die Änderung A11:2021 aktualisiert. Durch ISO 13485/A11:2021 wurde der Hauptteil der ISO 13485 nicht geändert. Neu hinzugekommen ist vor allem ein offizielles Mapping zur MDR. Dieses findet sich im Annex ZA.
Dieses Mapping ist bei der Analyse und Identifikation offener Anforderungen sehr hilfreich und sollte von Herstellern daher berücksichtigt werden.
Das heißt zusammengefasst, dass die Änderung A11:2021 keine neuen oder geänderten Anforderungen an das Qualitätsmanagementsystem stellt, sondern vor allem bei der Erlangung der MDR-Konformität unterstützt.
Da die neue Version harmoniert ist, sollten sie ab sofort die EN ISO 13485:2016/A11:2021 oder die DIN EN ISO 13485:2021 als Basis für Ihre MDR-Produkte verwenden.
2.6 QMS-Software, Atlassian und Co. – Das richtige Tooling
Sie sehen schon, ein Qualitätsmanagementsystem besteht aus zahlreichen Dokumenten und Aufzeichnungen, die in wechselseitiger Beziehung zueinander stehen. Ein sehr vernetztes System also, welches durch entsprechendes Tooling übersichtlicher gestaltet werden kann.
Qualitätsmanagementsystem klingt für viele Menschen immer noch nach sehr viel Papier. Natürlich kann man mit ausgedruckten Dokumenten arbeiten, und diese zum Beispiel mittels einer Unterschrift freigeben.
In der Regel ist dies aber nicht das Mittel der Wahl – gerade Software-Unternehmen, deren Mitarbeitende nicht immer an einem zentralen Ort arbeiten, stoßen mit diesem Ansatz auf Probleme.
Natürlich gibt es mittlerweile auch zahlreiche digitale Möglichkeiten, ein Qualitätsmanagementsystem umzusetzen. Ein paar Möglichkeiten zur Umsetzung sind in der untenstehenden Tabelle aufgeführt.
| Methode | Vorteile | Nachteile | Kommentar |
| Ausgedruckte Dokumente (z.B. Microsoft Word) |
|
|
Diese Methode ist besonders in älteren Unternehmen verbreitet. Vor allem in Software Unternehmen ist sie aber in den allermeisten Fällen nicht das Mittel zur Wahl. |
| Spezial-Software für QMS (z.B. Greenlight Guru) |
|
|
Spezial-Software kann in einigen Fällen sinnvoll sein – vor allem dann, wenn die Kosten erstmal keine große Rolle spielen. |
| Cloud-basierte Dokumentenmanagement-Software (z.B. Confluence/Jira) |
|
|
Diese Methode ist sehr flexibel und im Regelfall um einiges günstiger, als Spezial-Software für QMS. Daher ist sie besonders für Startups interessant. |
| Texteditor mit digitaler Versionsverwaltungs-Software (z.B. Word + Git) |
|
|
Auch diese Methode ist günstig und gut individualisierbar. Jedoch erfordert sie ein größeres technisches Verständnis als die anderen Methoden. |
3. Frequently asked questions (FAQ)
3.1 Wer braucht die ISO 13485?
Die ISO 13485 wird von Medizinproduktherstellern verwendet und beinhaltet Anforderungen an ein Qualitätsmanagementsystem. Ein solches wird beispielsweise von der MDR gefordert, um Medizinprodukte entwickeln zu können.
3.2 Was bestätigt ein ISO 13485 Zertifikat?
Ein ISO 13485 Zertifikat bestätigt, dass ein Unternehmen über ein Qualitätsmanagementsystem für Medizinprodukte verfügt. Ein solches ist in der gesetzlich vorgeschrieben, wenn Sie Medizinprodukte in der EU (und in vielen anderen Ländern) herstellen und vertreiben möchten. Es ist aber kein Nachweis dafür, dass Sie alle Anforderungen der MDR einhalten.
3.3 Ist die ISO 13485 Pflicht?
Nein, die ISO 13485 ist lediglich ein anerkannter Standard für Qualitätsmanagementsysteme für Medizinprodukte. Wer jedoch Medizinprodukte unter der MDR entwickeln möchte, ist mit einer entsprechenden Zertifizierung gut beraten. Vor allem eine benannte Stelle wird Ihr Qualitätsmanagementsystem unter die Lupe nehmen und ohne Zertifizierung könnte die Argumentation für Sie schwierig werden. Auch bei Herstellerüberwachungen von Aufsichtsbehörden ist ein ISO 13485 Zertifikat hilfreich.
3.4 Was muss ich bei Produkten mit künstlicher Intelligenz (KI) beachten?
Falls Sie künstliche Intelligenz (KI) in Ihr Produkt integrieren möchten, sollten einige weitere Aspekte bei der Umsetzung der ISO 13485 beachten. In unserem umfangreichen Leitfaden zu diesem Thema erklären wir im Detail, was für Sie bei der Zertifizierung von KI-Produkten besonders relevant ist: Zum KI-Leitfaden
3.5 Wie ist die Beziehung zwischen der ISO 13485 und der MDR?
Nach MDR müssen alle Medizinprodukt-Hersteller über ein Qualitätsmanagementsystem verfügen. Wie genau dieses auszusehen hat, spezifiziert die MDR allerdings nicht. Daher wurde die ISO 13485 zum Industriestandard in Europa, wenn es um Anforderungen an Qualitätsmanagementsysteme in der Medizinprodukt-Branche geht. Die ISO 13485 ist natürlich kein 1:1 Umsetzungsleitfaden für die MDR, denn es gibt auch Forderungen, die aus der MDR direkt hervorgehen und von der ISO 13485 nicht direkt abgedeckt werden (Beispielsweise die Ernennung einer “Person responsible for regulatory compliance – PRRC”, oder ein Prozess für die Meldung von Vorkommnissen an zuständige Behörden).
Wichtig: Die MDR spricht an keiner Stelle von der Umsetzung der ISO 13485, sondern immer von “Qualitätsmanagementsystem”. Formell gibt es also keine Pflicht, die ISO 13485 einzuhalten. Eine entsprechende Zertifizierung ist aber ein starker Nachweis für die Konformität Ihres Qualitätsmanagementsystems, weshalb wir eine solche sehr empfehlen. Die meisten MDR-Auditoren erwarten dies auch.
3.6 Kann die Herstellung von Medizinprodukten ausgelagert werden?
Ja, wenn Sie sich z. B. auf den Vertrieb und das Marketing fokussieren möchten, können Sie die Herstellung des Medizinprodukts auch an einen Dienstleister – wie QuickBird Medical – auslagern. Dadurch übernimmt der Dienstleister die Rolle des Inverkehrbringers und bringt das Medizinprodukt rechtlich auf den Markt. Somit geht die regulatorische Verantwortung auf den Dienstleister über.
Risikofreie Medizinprodukt-Zertifizierung ohne internen Aufwand für Ihr Team
Als zertifizierter Inverkehrbringer übernehmen wir für Sie die Hersteller-Verantwortung für Ihre Medizinprodukt-Software, Medical App oder DiGA. So müssen Sie kein Qualitätsmanagementsystem (QMS) bei sich aufbauen und werden als Unternehmen nicht durch die Regulatorik ausgebremst.
3.7 ISO 13485 als PDF herunterladen?
Die ISO 13485 ist kostengünstig über https://www.evs.ee/en zu erwerben und kann als PDF heruntergeladen werden (auf Englisch). Natürlich erhalten Sie die Norm auch auf zahlreichen anderen Plattformen, jedoch üblicherweise zu höheren Preisen. Die deutsche Version der ISO 13485 finden Sie unter anderem bei Beuth: https://www.beuth.de/de/norm/din-en-iso-13485/332674603
4. Weitere relevante Normen für Software-Hersteller
Als Ergänzung zur ISO 13485 müssen Sie sich als Hersteller von Software-Medizinprodukten auch mit weiteren Normen beschäftigen und diese umsetzen. Allen voran ist hier die IEC 62304 zu nennen, welche Anforderungen an die Software-Lebenszyklus-Prozesse stellt. Genaueres erfahren Sie in diesem Artikel: IEC 62304: Software-Lebenszyklus-Prozesse von Medizinprodukten
. Besonders für Hersteller von Standalone-Software lohnt sich außerdem ein Blick in die IEC 82304. Hier finden sich zwar zahlreiche Verweise auf die vorhin genannte IEC 62304, jedoch enthält sie zusätzliche Anforderungen an die Validierung von Gesundheitssoftware.
Zudem wird Ihnen auffallen, dass an einigen Stellen auf das Risikomanagement als begleitender Prozess verwiesen wird. Einen entsprechenden Prozess setzen Sie am besten nach ISO 14971 um, welcher durch die IEC 62366 (Anwendung der Gebrauchstauglichkeit auf Medizinprodukte) ergänzt wird. Weitere Informationen finden Sie in diesem Artikel: Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten
Natürlich gibt es noch andere Normen, die für Sie relevant sein könnten. Unsere Auflistung beinhaltet lediglich jene, welche aus unserer Sicht den größten Implementierungsaufwand benötigen.
5. Medizinprodukt-Zulassung ohne Qualitätsmanagement-System?
Gibt es einen Weg, die regulatorischen Pflichten für Medizinprodukt-Hersteller komplett zu vermeiden?
Ja, den gibt es: die Auslagerung der sogenannten Legalhersteller-Rolle an ein externes Unternehmen. Dies ist eine effiziente und risikominimierende Lösung, um Ihre Ressourcen zu schonen und gleichzeitig regulatorische Sicherheit zu schaffen. Sie müssen kein Qualitätsmanagement-System aufbauen und können den regulatorischen Zulassungsprozess vollständig an uns abgeben.
So können Sie sich auf die Produktkonzeption und Vermarktung konzentrieren, während QuickBird Medical als Legalhersteller alle regulatorischen Anforderungen übernimmt und für die Einhaltung haftet.
Mehr Informationen dazu finden Sie hier: Zum Fachartikel
6. Fazit
Die ISO 13485 ist die wohl verbreitetste Norm für den Aufbau von Qualitätsmanagementsystemen für Medizinprodukt-Hersteller in Europa. Wenn Sie diese einhalten, decken Sie die meisten Anforderungen der MDR in Bezug auf Qualitätsmanagementsysteme ab.
Die Unterkapitel in den Kapiteln 4 bis 8 beschreiben jeweils einzelne Anforderungen, die von Ihnen als Hersteller umzusetzen sind und stellen somit bereits die ideale Basis für einen entsprechenden Fahrplan dar.
Auch wenn die ISO 13485 für die durchaus verständlich geschrieben ist, ist ihre Umsetzung nicht ganz selbsterklärend. Die Besonderheiten einzelner Unternehmen müssen stets berücksichtigt werden, weshalb es kein pauschales Qualitätsmanagementsystem gibt, welches alle Software-Unternehmen gleichermaßen anwenden können.
Dennoch ist es ratsam, sich entsprechende Vorlagen zu kaufen und diese anschließend anzupassen.
Zudem empfehlen wir, den Aufbau Ihres Qualitätsmanagementsystems gemeinsam mit einem erfahrenen Berater vorzunehmen. Entsprechende Software-Tools können Ihnen außerdem einen maßgeblichen Effizienz-Vorteil verschaffen.
Dennoch, der Aufbau eines Qualitätsmanagementsystems ist sehr zeitaufwändig und produziert Kosten. Daher kann es für Sie sinnvoll sein, die Rolle des Inverkehrbringers für Ihr Produkt (zunächst) an einen Dienstleister zu übergeben. Dieser übernimmt dann die regulatorische Verantwortung für Ihr Produkt und steht rechtlich für seine Konformität ein. In diesem Fall entgehen Sie der Pflicht, ein eigenes Qualitätsmanagementsystem aufzubauen.
Wenn Sie die Umsetzung eines Software-Medizinprodukts und/oder den Aufbau eines Qualitätsmanagementsystems planen, sprechen Sie uns gerne an. Wir unterstützen Sie bei Ihrem Vorhaben. Kontaktieren Sie uns jetzt.

