

Many in vitro diagnostic devices today consist not only of physical test kits, but also of complex software that calculates, interprets, or visualizes analysis results. As a result, more and more software is falling within the scope of the In Vitro Diagnostic Regulation (IVDR).
This regulation governs products that examine samples outside the body, such as blood, urine, or tissue samples. The term “in vitro” literally means “in glass” and describes precisely these examinations outside the human body.
This article is aimed at (prospective) manufacturers of IVD products that consist either entirely or partially of software. We explain when software falls under the In Vitro Diagnostic Regulation (IVDR), how qualification works, and what criteria are used for risk classification.
Based on our practical experience in customer projects for the implementation of IVDR software (and even IVD apps), we can say that classification is not always easy. We therefore break down the topic for you and divide it into practical sub-aspects. This gives you a clear basis for qualifying and classifying your product.
Table of contents
- 1. Qualification: Does your Software fall under the IVDR?
- 2. Classification: Which Risk Class does your Software fall into?
- 3. Who makes the final Decision on Qualification and Classification?
- 4. Similarities and Differences: MDR vs. IVDR
- 5. Conclusion
1. Qualification: Does your Software fall under the IVDR?
The qualification serves to determine whether your software is considered an in vitro diagnostic device and therefore falls under the IVDR. The basis for this is the intended purpose (more on this here) of your product.
For qualification, you should check the following three aspects for your product.
1.1 Aspect 1: Definition of an IVD according to IVDR
In summary, your software falls within the regulated scope of the IVDR if it meets the definition of an “in vitro diagnostic device” under IVDR Article 2(2). The wording of this definition is as follows:
“In vitro diagnostic medical device” means any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.
Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices;
Therefore, check carefully whether the intended purpose of your product is covered by this definition.
1.2 Aspect 2: Definition of a Medical Device according to MDR
The first sentence of the above definition shows that an IVD must also be a “medical device”:
“In vitro diagnostic medical device” means any medical device which is a reagent,[…]
The IVDR refers to the Medical Device Regulation (MDR) for the definition of a medical device. Therefore, in addition to the above definition, you must also compare the definition of a medical device with the intended purpose of your product.
Our guide explains exactly what the definition of a medical device is according to the MDR and how you can determine whether your product falls under this definition: Is your software a medical device?
Important: Although you fall under the definition of a medical device according to MDR, you only need to comply with IVDR for an in vitro diagnostic device. MDR and IVDR do not apply to your product at the same time.
1.3 Aspect 3: Embedded, Accessory or Standalone software?
In addition to dealing with the IVD definition, it is also important to understand the “regulatory architecture” of your overall product:
- Is the software co-certified as part of your hardware device?
- Is the software to be treated separately and therefore certified as a separate IVD product?
- Is the software an accessory to the actual IVD product (e.g., hardware)?
The answers to these questions have a significant impact on the approval process for your product and should therefore be determined at the start of development.
To do this, you must decide which of the following three categories your software component falls into:
| Category | Explanation | Effects |
| #1: Part of an IVD device | Software is permanently connected to an IVD device and controls it or influences its use. It performs functions that are necessary for the operation of the device. | The software and device are certified as a single medical device. |
| #2: Accessories for an IVD device | Software is not an IVD itself, but is used to support the function of an IVD. It enables or improves the use of another IVD. See the definition of “accessory” in the IVDR. | Accessories are classified separately, regardless of the actual IVD with which they are used. |
| #3: Standalone IVD software | Software operates independently of a physical device and independently generates diagnostic information based on sample or measurement data. | Software is certified as a separate IVD according to IVDR. |
1.4 Summary: Aspects of Qualification
So you need to find out:
- Is your software part of a device, accessory, or standalone software?
- Does your software fall under the definition of a medical device according to MDR?
- Does your software fall under the definition of an in vitro diagnostic device according to IVDR?
You should review these topics at the beginning of your project to assess the applicability of the IVDR to your product.
1.5 Step-by-step Guide to Qualification
The answer to the key questions above is not always entirely clear. For this reason, there are additional guidance documents on the IVDR (and MDR) that are official in nature.
With regard to the qualification of software in relation to the IVDR, you should take a look at “MDCG 2019-11.” Among other things, this attempts to simplify and clarify the decision-making process for software within the framework of the IVDR: Link to MDCG Guidance
Although MDCG guidance documents are not legally binding in the strict sense, they are usually used by notified bodies in practice to make such decisions in the context of certification.
We have translated the MDCG 2019-11 decision tree and simplified the wording somewhat for greater clarity:
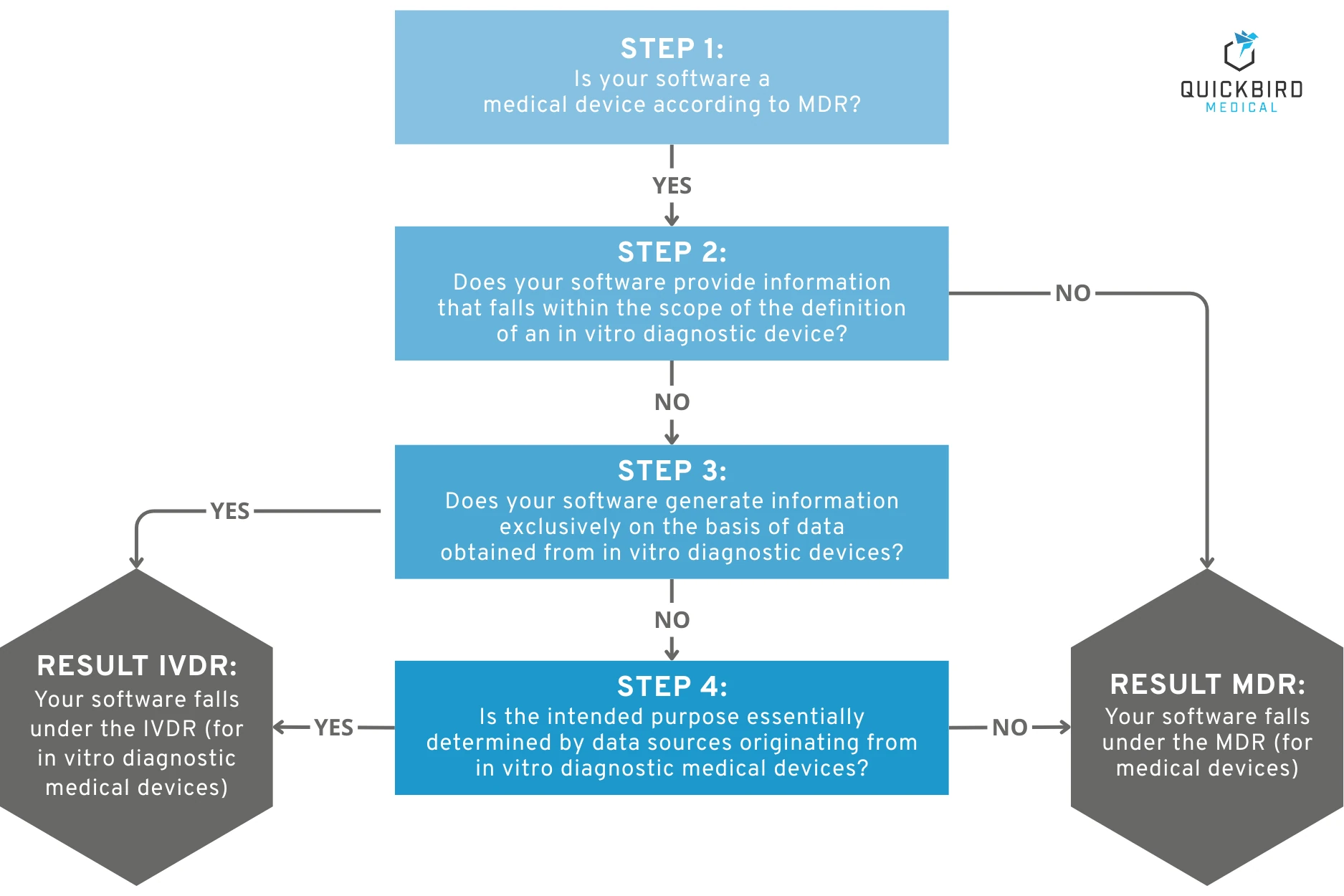
Decision tree for qualification: Does your software fall under the IVDR?
Step 1: Is your software a medical device according to MDR?
Does the intended purpose of your software fall under the definition of a medical device according to the MDR? If so, it is worth taking a look at our guide on this very topic: Guide: Is your software a medical device?
Step 2: Does your software provide information that falls within the scope of the definition of an in vitro diagnostic device?
Specifically: Does your software provide information for any of the following purposes? (see definition of an IVD in the IVDR):
a) physiological or pathological processes or conditions,
b) about congenital physical or mental impairments,
c) about the predisposition to a particular health condition or disease,
d) to determine the safety and tolerability in potential recipients,
e) about the expected effect of a treatment or the expected reactions to it, or
f) to determine or monitor therapeutic measures.
Step 3: Does your software generate information based solely on data obtained from in vitro diagnostic devices?
Are you sure that the data does not also originate from other sources, such as a medical device (not an IVD)? Only then can you answer this question with “Yes.”
Step 4: Is the intended purpose essentially determined by data sources originating from in vitro diagnostic medical devices?
If there is also information that does not originate from an IVD product, is the intended purpose still essentially determined by IVD information? To what extent does the “non-IVD information” influence the intended purpose?
Result: Have you reached the “IVDR result” at the bottom of the decision path?
Then your software is most likely an IVD or part of an IVD that falls under the IVDR. Unfortunately, every question and every definition is open to different interpretations. Therefore, you should not make this decision hastily and, if in doubt, seek professional advice on this matter.
Note: Our graphic is for illustrative purposes only. For the final qualification of the product, we recommend that you work with the English version of the MDCG Guidance in its original wording. Please feel free to contact us if you need input for the qualification or classification of your product. We develop IVDR and MDR software on a contract basis and are happy to share practical insights.
1.6 Qualification: Examples for IVDR and non-IVDR
To make the topic a little more tangible, here are some examples of software products that, according to MDCG guidance, tend to…
- … are an IVD according to IVDR.
- … are not IVDs, but are medical devices according to MDR.
- … are neither IVDs nor medical devices.
Qualification example: IVD according to IVDR
A clear example of an IVD is software that automatically evaluates raw data from laboratory tests. For example:
- Software that analyzes the optical density of an ELISA reader. In an ELISA, a chemical reaction in the sample causes a color change. The device sends light through the sample and measures how much light is absorbed. The color intensity can be used to determine how much of a particular substance is present in the blood.
- Software that interprets patterns from a so-called blot. A blot is a laboratory test in which proteins are transferred to a membrane, where they form visible lines or dots. These patterns indicate whether certain antibodies or proteins are present.
Such software records the measurement data and uses medical algorithms to calculate a diagnostic or prognostic finding. It is therefore generally considered to be IVD software.
Another exciting example that we at QuickBird Medical are very familiar with is Roche’s Accu-Chek SugarView app. The Accu-Chek SugarView app determines blood sugar levels by analyzing the Accu-Chek Active test strip and two photos taken of the test strip with a smartphone camera. The intelligent software works without any additional glucose meter. It has been classified as IVD software and has received CE marking (more information can be found here).
Qualification example: Not an IVD, but a medical device according to MDR
There is also software that serves a medical purpose but does not evaluate laboratory samples. Such applications are not covered by the IVDR but by the MDR. These include, in particular, programs that:
- Process or analyze medical images, for example in radiology or dermatology
- Evaluate measurements from a medical device, as long as these measurements do not originate from in vitro tests.
If the software does not examine blood, urine, or tissue samples but still supports medical decisions, then it may be a medical device according to the MDR and not an IVD.
Qualification example: No IVD and no MDR medical device
Many software products do not meet the definition of an IVD according to IVDR or a medical device according to MDR. These include, for example:
- Laboratory information systems (LIS) that only manage data or organize laboratory processes
- Programs that present results in a simple way, for example in the form of diagrams or simple statistics
- Software that merely stores or forwards measurement data without interpreting or analyzing it
Such software does not make its own diagnostic statements and has no direct medical purpose. Therefore, it does not fall under either the IVDR or the MDR.
2. Classification: Which Risk Class does your Software fall into?
“Yes, I do!” – Can you say this with confidence in relation to IVD qualification (see above)? So, do you fall within the scope of the IVDR? (For many manufacturers, this is probably not a case of “I do,” but rather “Yes, I have to 😔”). Then it’s time to take a look at the IVDR risk classes.
In order to determine the regulatory requirements for you as a manufacturer and your product, you first need to know which risk class your product or the components of your product fall into.
2.1 What Risk Classes are there under IVDR?
The IVDR distinguishes between the following risk classes: A, B, C, D
The idea is that products with a higher risk (e.g., B, C, or D) are also subject to stricter requirements than products with a very low risk (class A).
Note: This means that the risk classification under IVDR differs from that of medical devices covered by the Medical Device Regulation (MDR). The MDR divides medical devices into risk classes I, IIa, IIb, and III. For more information,
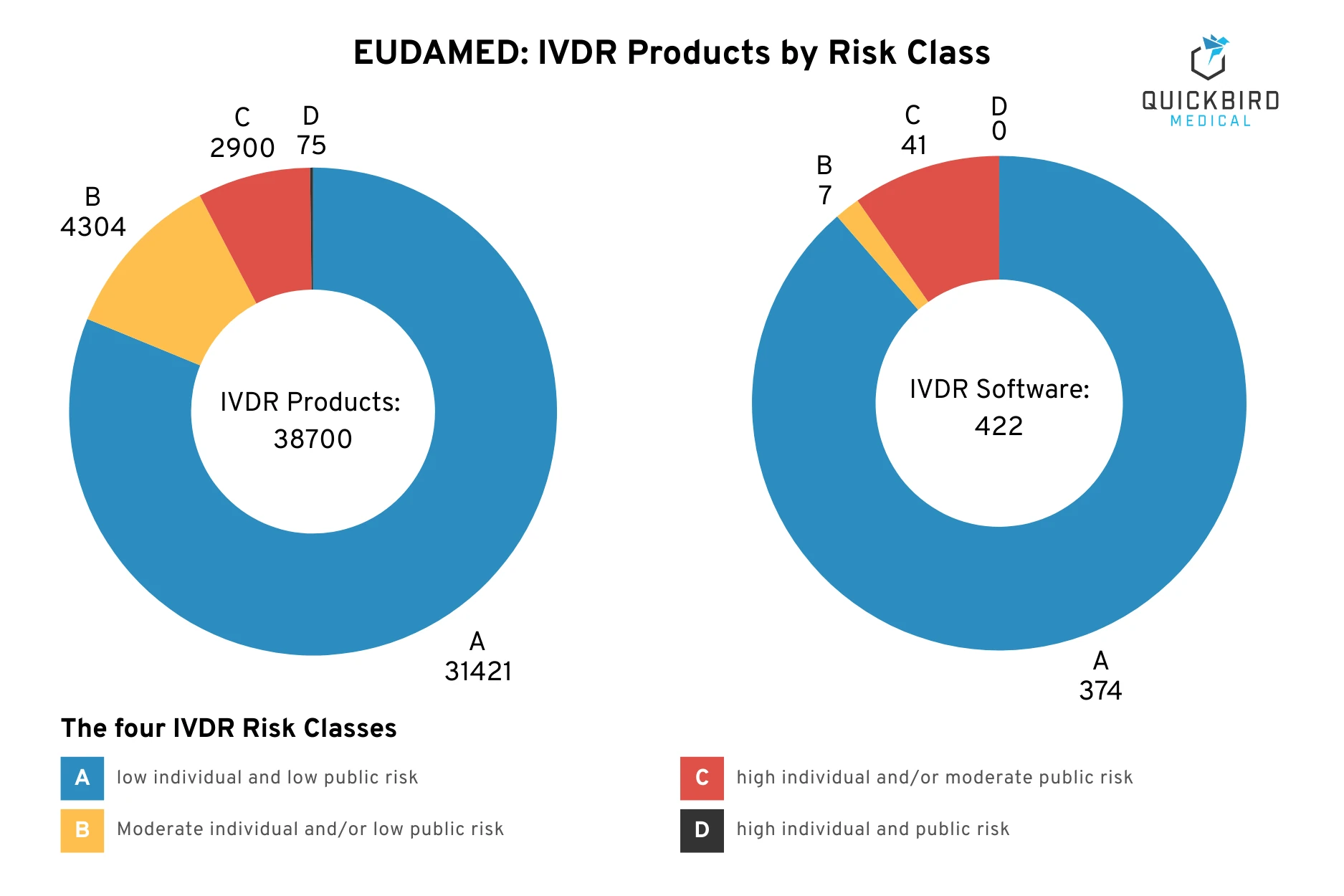
Search in the EUDAMED database: Number of IVDR products & IVDR software products per risk class
(Description of risks according to IMDRF guidance)
The figure shows the results of our research on the risk class distribution of IVDR products within the EUDAMED database. Please note: These statistics are not relevant to your product. There are many reasons why most products are classified in class A. Nevertheless, you must evaluate in a structured manner which risk class your software falls into (and in fact, especially in the case of diagnostic software, it is probably not class A or B).
2.2 The Main Implications of the IVDR Risk Classes
The risk class of an IVD software product affects development, the approval process, and post-market activities in many ways. Here we summarize the key aspects for you.
Identical requirements for quality management & technical documentation
Depending on the risk class, differently stringent requirements are imposed under the IVDR. However, this does not actually affect the so-called essential safety and performance requirements for the IVD product. The essential requirements for the manufacturer’s quality management system and the technical documentation of the product are identical for all risk classes (A, B, C, and D) (although additional requirements apply in isolated cases for the higher risk classes C & D).
The following are particularly relevant in this regard:
- Quality management according to ISO 13485 → Our guide to ISO 13485, specifically for software products
- Software lifecycle according to IEC 62304 → Our guide to IEC 62304
- Risk management according to ISO 14971
- Suitability for use according to IEC 62366-1
Notified body from risk class B onwards
Probably the biggest difference is that manufacturers of Class B, C, or D products (unlike most Class A products) are required to obtain a Certification process with a notified body that must be completed before the products can be placed on the market. An accredited body must therefore review and approve the technical documentation and the quality management system, among other things.
Incidentally, this is very similar to the regulation under the Medical Device Regulation (MDR), where Class I products are essentially certified by the manufacturer itself and can be placed on the market without external testing (in contrast to Classes IIa, IIb, and III under MDR).
Differences between classes B, C, and D
The difference between risk classes A and B is therefore clear (the assessment by the notified body). However, are there no further requirements above risk class B as the risk class increases (C and D)? Why then are there four classes and not just one?
Answer: Yes, there are. Put simply, for risk classes C and D:
- In some cases, additional documentation requirements are imposed (e.g., an additional brief report on safety and performance).
- The level of detail of the inspection by the notified body increases with the risk class.
- For risk class D, a European Union reference laboratory is involved in the testing and approval process.
You can also get a good overview by searching for the keyword “class” in the official legal text and checking all instances where it is mentioned.
2.3 Classification into the correct Class: IVDR Rules
First and foremost, it is important that the entire classification is based on the intended purpose of your product. We explain exactly how to define the intended purpose of a medical device in our separate guide here.
The classification of IVDR products is determined by a surprisingly clear, short list of seven rules (ANNEX VIII of the IVDR). Instead of repeating them here in a few words, we strongly recommend that you simply read the short section in the IVDR yourself (see below). We have included the exact wording from the regulation for you in the next chapter. This is actually the only section of the IVDR that deals with classification into risk classes.
2.3.1 Excerpt from the EU Regulation: Classification rules according to Annex VIII of the IVDR
The following classification rules can be found in the In Vitro Diagnostic Medical Devices Regulation (IVDR) in Annex VIII:
2. CLASSIFICATION RULES
2.1 Rule 1
Devices intended to be used for the following purposes are classified as class D:
- detection of the presence of, or exposure to, a transmissible agent in blood, blood components, cells, tissues or organs, or in any of their derivatives, in order to assess their suitability for transfusion, transplantation or cell administration;
- detection of the presence of, or exposure to, a transmissible agent that causes a life-threatening disease with a high or suspected high risk of propagation;
- determining the infectious load of a life-threatening disease where monitoring is critical in the process of patient management.
2.2. Rule 2
Devices intended to be used for blood grouping, or tissue typing to ensure the immunological compatibility of blood, blood components, cells, tissue or organs that are intended for transfusion or transplantation or cell administration, are classified as class C, except when intended to determine any of the following markers:
- ABO system [A (ABO1), B (ABO2), AB (ABO3)];
- Rhesus system [RH1 (D), RHW1, RH2 (C), RH3 (E), RH4 (c), RH5 (e)];
- Kell system [Kel1 (K)];
- Kidd system [JK1 (Jka), JK2 (Jkb)];
- Duffy system [FY1 (Fya), FY2 (Fyb)]; in which case they are classified as class D. L 117/304 EN Official Journal of the European Union 5.5.2017
2.3. Rule 3
Devices are classified as class C if they are intended:
(a) for detecting the presence of, or exposure to, a sexually transmitted agent;
(b) for detecting the presence in cerebrospinal fluid or blood of an infectious agent without a high or suspected high risk of propagation;
(c) for detecting the presence of an infectious agent, if there is a significant risk that an erroneous result would cause death or severe disability to the individual, foetus or embryo being tested, or to the individual’s offspring;
(d) for pre-natal screening of women in order to determine their immune status towards transmissible agents;
(e) for determining infective disease status or immune status, where there is a risk that an erroneous result would lead to a patient management decision resulting in a life-threatening situation for the patient or for the patient’s offspring;
(f) to be used as companion diagnostics;
(g) to be used for disease staging, where there is a risk that an erroneous result would lead to a patient management decision resulting in a life-threatening situation for the patient or for the patient’s offspring;
(h) to be used in screening, diagnosis, or staging of cancer;
(i) for human genetic testing;
(j) for monitoring of levels of medicinal products, substances or biological components, when there is a risk that an erroneous result will lead to a patient management decision resulting in a life-threatening situation for the patient or for the patient’s offspring;
(k) for management of patients suffering from a life-threatening disease or condition; (l) for screening for congenital disorders in the embryo or foetus;
(m) for screening for congenital disorders in new-born babies where failure to detect and treat such disorders could lead to life-threatening situations or severe disabilities.2.4. Rule 4
(a) Devices intended for self-testing are classified as class C, except for devices for the detection of pregnancy, for fertility testing and for determining cholesterol level, and devices for the detection of glucose, erythrocytes, leucocytes and bacteria in urine, which are classified as class B.
(b) Devices intended for near-patient testing are classified in their own right.2.5. Rule 5
The following devices are classified as class A:
(a) products for general laboratory use, accessories which possess no critical characteristics, buffer solutions, washing solutions, and general culture media and histological stains, intended by the manufacturer to make them suitable for in vitro diagnostic procedures relating to a specific examination;
(b) instruments intended by the manufacturer specifically to be used for in vitro diagnostic procedures;
(c) specimen receptacles.2.6. Rule 6
Devices not covered by the above-mentioned classification rules are classified as class B.
2.7. Rule 7
Devices which are controls without a quantitative or qualitative assigned value are classified as class B.
2.3.2 Excerpt from the EU Regulation: Implementing Rules for Classification
In addition to the classification rules, the IVDR also contains a number of implementing provisions relating to classification. Below you will find an excerpt from Annex VIII of the IVDR.
1. IMPLEMENTING RULES
1.1. Application of the classification rules shall be governed by the intended purpose of the devices.
1.2. If the device in question is intended to be used in combination with another device, the classification rules shall apply separately to each of the devices.
1.3. Accessories for an in vitro diagnostic medical device shall be classified in their own right separately from the device with which they are used.
1.4. Software, which drives a device or influences the use of a device, shall fall within the same class as the device. If the software is independent of any other device, it shall be classified in its own right.
1.5. Calibrators intended to be used with a device shall be classified in the same class as the device.
1.6. Control materials with quantitative or qualitative assigned values intended for one specific analyte or multiple analytes shall be classified in the same class as the device.
1.7. The manufacturer shall take into consideration all classification and implementation rules in order to establish the proper classification for the device.
1.8. Where a manufacturer states multiple intended purposes for a device, and as a result the device falls into more than one class, it shall be classified in the higher class.
1.9. If several classification rules apply to the same device, the rule resulting in the higher classification shall apply.
1.10. Each of the classification rules shall apply to first line assays, confirmatory assays and supplemental assays.
2.4 MDCG Guidance on Classification under IVDR
Not every sentence in the IVDR classification rules shown above is self-explanatory and unambiguous. Therefore, there are additional MDCG guidelines to help you determine the risk class, among other things.
There are two relevant MDCG guidance documents for the classification of software products:
- General Guidance – “MDCG 2020-16: Guidance on Classification Rules for in vitro Diagnostic Medical Devices under
Regulation (EU) 2017/746: This document provides helpful examples and comments on each classification rule in the IVDR. This gives you additional support in evaluating whether certain classification rules apply to your product. –> Link - Software-specific for MDR and IVDR – “MDCG 2019-11: Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 IVDR”: Among other things, the document addresses the specifics of software qualification and classification. In contrast to MDCG 2020-16, the scope of the relevant sections is relatively brief. –> Link
Note: The term “Regulation (EU) 2017/746” refers to the IVDR. “Regulation (EU) 2017/745,” on the other hand, refers to the MDR.
2.5 Classification: Software Examples for all Classes of the IVDR
We would also like to provide you with some software examples from the MDCG Guidance to illustrate the topic of classification. Of course, these are only illustrations, and sometimes the devil is in the details. In this respect, you should not classify your product purely on the basis of a comparison with existing examples, but should check all the rules of the IVDR individually.
Software example for risk class A
Software belongs primarily to risk class A if it is intended exclusively to control an IVD device (according to IVDR) that is also classified in class A. This is stated in Annex VIII 1.4 of the IVDR: “Software that controls a product or influences its use shall be classified in the same class as the product.”
A concrete example is software that remotely operates a CRP analyzer (C-reactive protein device), which falls under Class A. The software does not make any diagnostic statements of its own, but only ensures that the device functions. It is therefore classified in the same class as the device, in this case Class A.
Software example for risk class B
It is somewhat difficult to find official examples of Class B software. (Software) products only end up in Class B if they fall into specific product categories for self-use (e.g., pregnancy tests) (Rule 4) or if none of the other classification rules apply (Rule 6).
The following example can be found in Guidance MDCG 2020-16:
„Blood gas analyser and the software driving and influencing it are classified as […] Class B (Rule 6) where the blood gas analyser directly measures a parameter from a specimen (in the absence of a reagent or disposable sensor cassette) AND where an erroneous result in a measured parameter WILL NOT LEAD TO a patient management decision resulting in a lifethreatening situation.“
Software example for risk class C
Software that calculates HbA1c values in an automated ELISA system and is used for diabetes diagnosis or monitoring falls under class C. This can be inferred from Rule 3(k) of the IVDR.
Software that classifies smears as abnormal or normal in an automated PAP screening system also falls under Class C. This can be inferred from Rule 3(h) of the IVDR.
Software example for risk class D
Software that interprets the results of an automated line immunoassay for HIV antibodies falls under class D. This can be derived from Rule 1 of the IVDR. In this case, the software evaluates highly critical infection markers.
3. Who makes the final Decision on Qualification and Classification?
As the manufacturer of the product, you must make this decision. The responsibility for checking the applicability of the IVDR to your (software) product (qualification) and classifying it into the appropriate risk class lies entirely with you initially.
Careful assessment is crucial here, because a notified body can refuse certification if it comes to a different conclusion in its risk classification. In addition, you are of course legally vulnerable if you do not carry out this classification correctly.
You should be particularly cautious when classifying a product in risk class A, as the classification is not initially checked by a notified body. This means you have even less perceived security when selecting the risk class.
Furthermore, although no notified body is involved here, the competent supervisory authority may notice an incorrect classification and, in the worst case, demand that the product be withdrawn from the market. Even products in risk class A are subject to irregular inspections. Furthermore, in the event of personal injury caused by your product, there will certainly be legal questions as to whether the risk class assigned by you as the manufacturer was appropriate.
If you are unsure, it is worth obtaining an independent assessment. Possible points of contact are:
- a specialized lawyer who can prepare an expert opinion
- A regulatory expert or consultant with relevant experience
- the BfArM (in practice, however, difficult due to months-long processing times)
QuickBird Medical develops IVDR and MDR software for customers on a contract basis. We are happy to provide you with input on classification and qualification decisions based on our experience. If necessary, we can also refer you to a lawyer from our network who can prepare an expert opinion for you.
→ Contact us
Important: Neither an external assessment nor a formal expert opinion offers complete legal protection. However, they can serve as an important basis for decision-making in the event of a legal dispute.
4. Similarities and Differences: MDR vs. IVDR
The MDR regulates traditional medical devices, which are usually used directly on patients, while the IVDR applies to in vitro diagnostic medical devices, i.e., products that examine samples such as blood or tissue.
Both regulations function similarly in many areas: manufacturers need a quality management system, technical documentation, a person responsible for regulatory compliance, and must classify their products according to fixed risk classes.
Major differences include the following points:
- Different classification logic: The risk classes and classification logic of the IVDR and MDR differ significantly. It should also be noted here that software products under the IVDR fall into the lowest class (A) much less frequently than is the case with class I under the MDR. This means that a notified body is involved in most IVD products with software.
- More specific performance evaluation for IVDs: IVD products must undergo a very comprehensive performance evaluation. This includes scientific validity, analytical performance (e.g., sensitivity, specificity), and clinical performance. Under the MDR, there are also medical devices that must comply with such validation requirements (e.g., a product for diagnosing diseases based on CT image data). However, these requirements are defined much more specifically in the IVDR than in the MDR.
- Additional testing mechanisms for high-risk IVDs: IVDR Class D products (e.g., HIV or hepatitis tests) must be independently tested by EU reference laboratories (EURLs).
5. Conclusion
IVDR software operates in the area of conflict between technical development, laboratory reality, and strict regulatory requirements.
This article is intended to help you answer the first key questions: Does your software fall under the IVDR at all, and which risk class does it belong to? Once these cornerstones are in place, you will have a solid basis for tackling quality management, further documentation, validation, and cooperation with a notified body in a targeted manner.
In the context of the IVDR, it should be noted that the majority of all software-based diagnostic products will not fall into risk class A. Therefore, as a manufacturer, you should expect to have to include a notified body in the approval process. You should take the costs and additional time required for this certification into account in your product planning.
About us: QuickBird Medical develops IVDR and MDR software for customers on a contract basis. We take care of both the technical implementation and regulatory compliance of your (software) product. So if you are planning an IVDR or MDR product that includes software components, please feel free to contact us for a no-obligation initial consultation.
→ Contact us
In the future, we plan to publish further articles to assist you in the planning and development of IVDR products, specifically the software components. Our newsletter will keep you informed about new articles and changes related to this topic.

