

Although Switzerland is geographically located in the heart of the EU, it is not part of it. This also affects manufacturers of (software) medical devices.
Is it possible for MDR medical devices to enter the Swiss market without further ado? How do Swiss manufacturers enter the EU market with their products? And what do you need an authorized representative for?
In this article, we clarify these and other questions about entering the other economic area: from the EU to Switzerland and vice versa.
We focus here on software medical devices, but the content also applies to other types of medical devices.
Contents
- Swiss Medical Devices Ordinance (MedDO) vs MDR
- Entering the Swiss market as an EU manufacturer
- Entering the EU market as a Swiss manufacturer
- Conclusion
Note: As we cannot go into every special case in this article, the requirements that generally apply to manufacturers are described here. However, we advise you to contact the competent supervisory authority to obtain information on any transitional regulations.
Swiss Medical Devices Ordinance (MedDO) vs MDR
To begin with, we need to look at the legal framework in Switzerland and the EU to understand what manufacturers have to comply with in each region.
The Swiss Medical Devices Ordinance (MedDO) is the applicable law for medical devices in Switzerland. At first it sounds as if there are completely different regulations to those defined by the MDR. On closer inspection, however, there are strong parallels, numerous references and even complete copies of the MDR in the MedDO.
So there’s no need to panic – we summarize the most important differences and the implications for you as a manufacturer in this article.
Definition of medical device
The definition of a medical device is almost identical in the MDR and MedDO – apart from a few differences in wording. This means that there is no additional work for EU and Swiss manufacturers in the area of qualification if they wish to expand their market to the EU or Switzerland.
If you do not yet know whether your product is a medical device, read our guide: Is my app a medical device?
EU-Switzerland Framework Agreement
A valid framework agreement between Switzerland and the EU was discussed for a long time, but unfortunately failed in the end. A detailed discussion of the reasons for this does not provide any real added value for you as a manufacturer, so we will not discuss this topic further. The only important thing at this point is:
- Switzerland is considered a third country within the meaning of the MDR.
This means even more concretely:
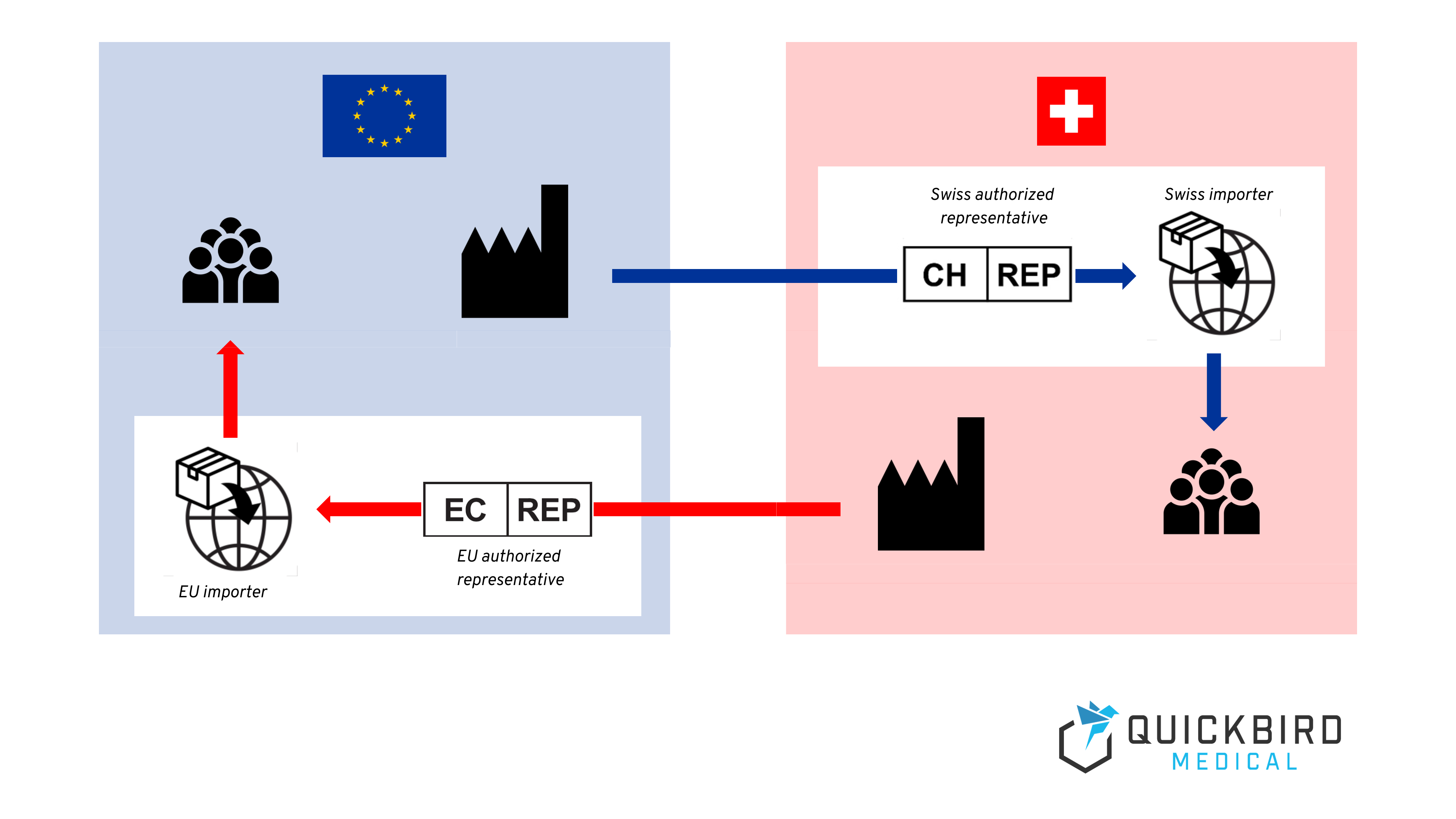
- You need an authorized representative based in Switzerland or the EU to be able to place a medical device on the respective market (unless you have your own registered office there).
Entering the Swiss market as an EU manufacturer
If you are a medical device manufacturer based only in the EU and do not have a registered office in Switzerland, your products will certainly have MDD or MDR approval. There is good news at this point: Products bearing a CE marking may also be placed on the market in Switzerland and no additional certification obligations apply.
Swissmedic (the Swiss regulatory and supervisory authority for medicinal products and medical devices) explicitly states on its website that products with a CE marking can be placed on the market in Switzerland. Swissmedic’s main function would be to monitor medical devices rather than to authorize them.
Nevertheless, there are a few things you need to do to be able to sell your product legally in Switzerland:
- Appointment of an authorized representative in Switzerland
- Instruction of the authorized representative and clarification of duties
- Registration of the authorized representative and your product in the Swissmedic database
- Publication of summary reports on the safety and performance of devices (only for Class III and implantable devices)
- Designation of an importer in Switzerland
Authorized representative in Switzerland
If your company does not have its own registered office in Switzerland, you must appoint an authorized representative who will assume various obligations arising from the MedDO and MDR on your behalf. This representative must be a natural person or legal entity domiciled in Switzerland.
The authorized representative is responsible for formal and safety-related matters in connection with placing the product on the market. This includes, for example, the obligation to report incidents and trends, for which the authorized representative is always responsible under Swiss law (unlike in the MDR).
If you have read the obligations of the authorized representative in Article 51, MedDO you will quickly see that these are otherwise (almost) identical to those of the MDR. This is because it simply refers to Article 11 of the MDR.
Which tasks you delegate to the authorized representative is partly up to you, but there are restrictions here. Not all manufacturer obligations can be transferred to the authorized representative – this includes, for example, conducting a clinical evaluation or responsibility for risk management (all exceptions can be found in paragraph 4, Article 11 MDR).
Irrespective of this, the authorized representative is obliged to ensure that the authority receives the complete technical documentation for a product within seven days of a request from the authority. However, the Swiss authorized representative is not obliged to have a copy of this documentation itself – it can remain with the manufacturer.
Service providers who offer this service in Switzerland, for example, are suitable for the role of authorized representative.
Importer in Switzerland
In addition to an authorized representative, who assumes certain obligations in terms of medical device regulation, EU manufacturers also need an importer in Switzerland in order to place the product on the market there.
The MedDO defines the importer as “any natural or legal person established in Switzerland who places a product from abroad on the Swiss market;”
The latter has similar obligations to the authorized representative – these can be found in detail in Articles 13 and 16 of the MDR and Article 51 of the MedDO. In contrast to the authorized representative, however, this person does not need to have the necessary regulatory expertise in the field of medical devices. However, an importer also assumes certain obligations with regard to the storage and transportation of medical devices, for example – but since (pure) software is not a physical product, this requirement cannot usually be implemented in a meaningful way for software medical devices.
The authorized representative himself can also assume the role of importer. In this case, however, both roles must be registered in the Swissmedic database. You can find out more about this in the next chapter.
Registration in the Swissmedic database
Every economic operator (manufacturer, authorized representative and importer) must register in the Swissmedic database within 3 months of placing a medical device on the market in Switzerland. The name, address and contact details of the economic operator and the responsible person are required. After registration, the user receives a CHRN – this is a unique identifier. It is the equivalent of the SRN in EUDAMED.
As Swissmedic does not have access to EUDAMED, a separate reporting system for medical devices is to be set up in order to be able to monitor them. However, such a system is not yet available, which is why the registration of medical devices by filling out and submitting a form . The product must be registered at the latest when it is placed on the market.
Entering the EU market as a Swiss manufacturer
If you, as a Swiss manufacturer, want to launch a product on the EU market, we must first distinguish whether you already have a medical device or whether its development is still in progress. We examine both cases in the following chapters.
IMPORTANT: As a Swiss manufacturer without a registered office in the EU, you always need an authorized agent/representative and an importer based in the EU. You must also register your medical device in the EUDAMED database.
You do not yet have a medical device
When developing medical devices for the EU, the MDR applies first and foremost.
As the MDR is recognized in Switzerland and the MedDO refers to it in many places, the requirements for product development are largely identical. This includes, for example, the development of a quality management system and the creation of technical documentation. We have already summarized the exact requirements in other blog articles:
- Guidelines for the development of medical apps: what manufacturers need to look out for (MDR)
- Guide: Is your app a medical device?
The question of whether you want to enter the Swiss market (under MedDO) or the EU market (under MDR) first should be clarified at the outset. Both have certain advantages and disadvantages, which you need to weigh up for your company. Among other things, you need to find out which market is more interesting for you and to what extent you want to disclose your technical documentation (e.g. to an EU authorized representative).
If you have already developed and approved a medical device, continue reading in the next chapter.
You already have a medical device according to MedDO
If you have already developed a medical device that complies with the MedDO, you have also carried out a conformity assessment procedure for the product. Before you can place the product on the EU market, you must check whether the procedure carried out is also permitted for the EU. If a designated body was involved in the conformity assessment of the medical device, make sure that it is also recognized as a notified body according to MDR. In many cases, this is not the case and you must carry out a further conformity assessment procedure in accordance with the MDR.
You also need an authorized representative and an importer based in the EU and must register your product in EUDAMED. You can find out more about this in the following chapters.
Conformity assessment procedure according to MDR
In order to be able to affix the CE marking to a medical device, manufacturers must carry out a so-called conformity assessment procedure. The MDR offers three different assessment procedures, which are congruent with the procedures of the MedDO. For software products, this is usually an assessment based on the quality management system and the technical documentation. You can find more information on this topic in our article: Approval & certification of software medical devices (MDR)
Authorized representative & importer in the EU
Just like EU manufacturers in Switzerland, Swiss manufacturers in the EU also need an authorized representative (also known as a “representative”) to be able to sell their products on the EU market.
The authorized representative in the EU has the same obligations as his equivalent in Switzerland (see above) – with one exception:
The EU representative must have a copy of the technical documentation and the EU Declaration of Conformity and be able to provide these to the competent authorities on request.
Just as in Switzerland, manufacturers also need an importer within the EU to import the product into the EU and make it available on the market.
Are you a medical device manufacturer based in Switzerland? Then contact us – we will be happy to take on the role of authorized representative and importer and ensure the smooth marketing of your products in the EU.
Registration in EUDAMED
All economic operators (manufacturer, authorized representative and importer) must be registered in EUDAMED. This registration takes place via the corresponding online portal, after which you will receive an SRN (unique identifier).
Medical devices themselves must also be registered in the EUDAMED database. You can find out everything about the certification and approval of medical devices under MDR in our article: Approval & certification of software medical devices (MDR)
Conclusion
As there is currently no longer an agreement between Switzerland and the EU, Switzerland is treated as a third country for the purposes of the MDR. This means that medical device manufacturers need an authorized representative in the EU in order to place a product on the market there. The situation is the same the other way round, but medical devices with a CE marking are recognized unilaterally in Switzerland, so there are no additional certification obligations for EU manufacturers.
Swiss manufacturers, on the other hand, often have to carry out a further conformity assessment procedure (involving a notified body) in order to be able to offer their medical devices in the EU.
Based in Germany, QuickBird Medical is happy to take on the role of authorized representative and importer within the EU. Contact us if you need support with the placing on the market or the development of MDR medical devices. We will be happy to assist you further.

