

Die Schweiz befindet sich geografisch zwar im Herzen der EU, ist aber trotzdem kein Teil davon. Das hat auch Auswirkungen auf Hersteller von (Software-)Medizinprodukten.
Ist ein Markteintritt in die Schweiz für MDR-Medizinprodukte ohne Weiteres möglich? Wie kommen Schweizer Hersteller mit Ihren Produkten in den EU-Markt? Und wofür benötigt man einen Bevollmächtigten?
In diesem Artikel klären wir diese und weitere Fragen zum Eintritt in den jeweils anderen Wirtschaftsraum: von der EU in die Schweiz und andersherum.
Wir fokussieren uns hier auf Software-Medizinprodukte, die Inhalte treffen aber ebenso auf andere Arten von Medizinprodukten zu.
Inhalt
- 1. Schweizer Medizinprodukteverordnung (MepV) vs MDR
- 2. Als EU-Hersteller auf den Schweizer Markt kommen
- 3. Als Schweizer Hersteller auf den EU-Markt kommen
- 4. Fazit
Hinweis: Da wir in diesem Artikel nicht auf jeden Sonderfall eingehen können, werden hier die Anforderungen beschrieben, die im Allgemeinen für Hersteller gelten. Wir raten aber dazu, sich mit der zuständigen Aufsichtsbehörde in Verbindung zu setzen, um Auskunft zu etwaigen Übergangsregelungen zu erhalten.
1. Schweizer Medizinprodukteverordnung (MepV) vs MDR
Zu Beginn müssen wir uns den rechtlichen Rahmen in der Schweiz und der EU ansehen, um zu verstehen, woran sich Hersteller in den jeweiligen Regionen halten müssen.
Die schweizerische Medizinprodukteverordnung (MepV) ist geltendes Recht für Medizinprodukte in der Schweiz. Zunächst klingt das so, als gäbe es komplett unterschiedliche Regelungen, als jene von der MDR definierten. Bei genauerer Betrachtung finden sich jedoch starke Parallelen, zahlreiche Verweise und sogar komplette Kopien der MDR in der MepV wieder.
Kein Grund zur Panik also – die wichtigsten Unterschiede und die Implikationen, die sich daraus für Sie als Hersteller ableiten, fassen wir in diesem Artikel zusammen.
1.1 Definition Medizinprodukt
Die Definition eines Medizinprodukts ist in MDR und MepV nahezu deckungsgleich – bis auf ein paar unterschiedliche Formulierungen. Somit gibt es im Bereich der Qualifikation keinen Mehraufwand für EU- und Schweizer-Hersteller, wenn sie ihren Markt auf die EU bzw. Schweiz erweitern möchten.
Wenn Sie noch nicht wissen, ob es sich bei Ihrem Produkt um ein Medizinprodukt handelt, lesen Sie unseren Leitfaden: Ist meine App ein Medizinprodukt?
1.2 Rahmenabkommen EU-Schweiz
Ein geltendes Rahmenabkommen zwischen der Schweiz und der EU wurde lange diskutiert, ist am Ende aber leider gescheitert. Eine genaue Erörterung der Gründe dafür liefert für Sie als Hersteller keinen wirklichen Mehrwert, daher werden wir dieses Thema nicht weiter behandeln. Wichtig ist an dieser Stelle nur:
- Die Schweiz gilt als Drittland im Sinne der MDR.
Das bedeutet noch konkreter:
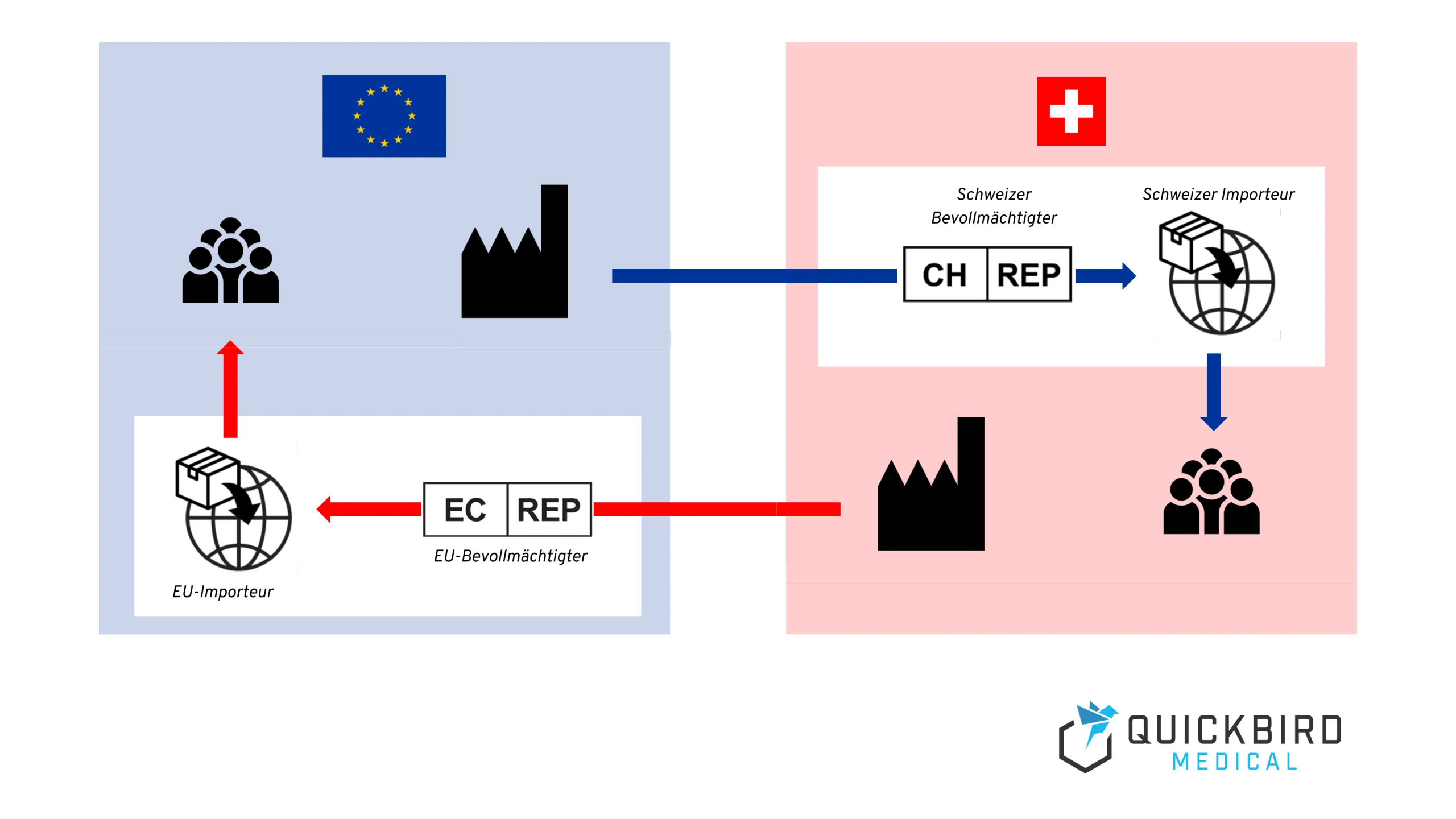
- Sie brauchen einen Bevollmächtigten mit Sitz in der Schweiz oder der EU, um ein Medizinprodukt auf den jeweiligen Markt bringen zu können (sofern Sie keinen eigenen Sitz dort haben).
Das Rahmenabkommen der EU mit der Schweiz vereinfacht dargestellt
2. Als EU-Hersteller auf den Schweizer Markt kommen
Wenn Sie als Medizinprodukt-Hersteller nur in der EU ansässig sind und keinen Sitz in der Schweiz haben, besitzen Ihre Produkte bestimmt eine MDD- oder MDR-Zulassung. An dieser Stelle gibt es gute Nachrichten: Produkte, die eine CE-Kennzeichnung tragen, dürfen auch in der Schweiz auf den Markt gebracht werden und es gelten keine zusätzlichen Zertifizierungspflichten.
Die Swissmedic (schweizerische Zulassungs- und Aufsichtsbehörde für Arzneimittel und Medizinprodukte) schreibt auf ihrer Webseite explizit, dass Produkte mit einer CE-Kennzeichnung in der Schweiz in Verkehr gebracht werden können. Die Hauptfunktion der Swissmedic läge insbesondere in der Überwachung von Medizinprodukten, weniger in deren Zulassung.
Dennoch gibt es ein paar Dinge, die Sie tun müssen, um Ihr Produkt legal in der Schweiz vertreiben zu können:
- Ernennung eines Bevollmächtigten in der Schweiz
- Weisung des Bevollmächtigten und Klärung der Pflichten
- Anmeldung des Bevollmächtigten und Ihres Produkts in der Swissmedic Datenbank
- Veröffentlichung von Kurzberichten zur Sicherheit und Leistungsfähigkeit von Produkten (nur für Klasse III- und implantierbare Produkte)
- Bestimmung eines Importeurs in der Schweiz
2.1 Bevollmächtigter in der Schweiz
Wenn Ihr Unternehmen keinen eigenen Firmensitz in der Schweiz hat, müssen Sie einen Bevollmächtigten ernennen, welcher diverse Pflichten aus der MepV bzw. MDR für Sie übernimmt. Dieser Repräsentant muss eine natürliche oder juristische Person mit Sitz in der Schweiz sein.
Der Bevollmächtigte ist zuständig für die formellen und sicherheitsrelevanten Belange im Zusammenhang mit dem Inverkehrbringen des Produkts. Dazu zählt beispielsweise die Pflicht zur Meldung von Vorkommnissen und Trends, für die nach schweizerischem Recht immer der Bevollmächtigte verantwortlich ist (anders als in der MDR).
Wenn Sie die Pflichten des Bevollmächtigten in Artikel 51, MepV nachlesen, sehen Sie schnell, dass diese ansonsten (fast) deckungsgleich mit jenen der MDR sind. Dort wird nämlich schlichtweg auf Artikel 11 der MDR verwiesen.
Welche Aufgaben Sie an den Bevollmächtigten delegieren, bleibt zum Teil Ihnen überlassen, allerdings gibt es hier Einschränkungen. Nicht alle Herstellerpflichten können an den Bevollmächtigten abgegeben werden – dazu zählt beispielsweise die Durchführung einer klinischen Bewertung oder die Verantwortung für das Risikomanagement (alle Ausnahmen finden Sie in Absatz 4, Artikel 11 MDR).
Unabhängig davon hat der Bevollmächtigte die Pflicht, bei einer Anfrage der Behörde innerhalb von sieben Tagen dafür zu sorgen, dass diese die vollständige technische Dokumentation zu einem Produkt erhält. Der Schweizer Bevollmächtigte ist allerdings nicht dazu verpflichtet, eine Kopie dieser Dokumentation selbst zu haben – diese kann beim Hersteller bleiben.
Für die Rolle des Bevollmächtigten eignen sich beispielsweise Dienstleister, die diesen Service in der Schweiz anbieten.
2.2 Importeur in der Schweiz
Zusätzlich zu einem Bevollmächtigten, welcher gewisse Pflichten im Sinne der Medizinprodukt-Regulatorik übernimmt, benötigen EU-Hersteller auch einen Importeur in der Schweiz, um das Produkt dort in Verkehr zu bringen.
Die MepV definiert den Importeur als “jede in der Schweiz niedergelassene natürliche oder juristische Person, die ein Produkt aus dem Ausland auf dem Schweizer Markt in Verkehr bringt;”
Dieser hat ähnliche Pflichten, wie der Bevollmächtigte – im Detail nachzulesen sind diese in Artikel 13 und 16 der MDR bzw. Artikel 51 der MepV. Im Unterschied zum Bevollmächtigten benötigt er aber keine Person mit dem notwendigen regulatorischen Fachwissen im Medizinprodukte-Bereich. Ein Importeur übernimmt zum Beispiel aber auch bestimmte Pflichten in Bezug auf die Lagerung und den Transport von Medizinprodukten – da es sich bei (reiner) Software aber um kein physisches Produkt handelt, ist diese Anforderung bei Software-Medizinprodukten in der Regel nicht sinnvoll umsetzbar.
Auch der Bevollmächtigte selbst kann die Rolle des Importeurs übernehmen. In diesem Fall sind allerdings beide Rollen in der Swissmedic Datenbank anzumelden. Mehr dazu erfahren Sie im nächsten Kapitel.
2.3 Anmeldung in der Swissmedic Datenbank
Jeder Wirtschaftsakteur (Hersteller, Bevollmächtigter und Importeur) muss sich innerhalb von 3 Monaten, nachdem ein Medizinprodukt in der Schweiz in Verkehr gebracht wurde, in der Swissmedic Datenbank anmelden. Dabei werden Name, Anschrift und Kontaktdaten des Wirtschaftsakteurs, sowie der verantwortlichen Person gefordert. Nach der Registrierung erhält dieser eine CHRN – dabei handelt es sich um eine eindeutige Kennung. Sie ist das Äquivalent zur SRN in EUDAMED.
Der deutsche DiGA-Fast-Track – ein Modell für ganz Europa?
In diesem Whitepaper sehen wir uns an, in welchen EU-Ländern bereits ein DiGA-Modell existiert, ein DiGA-Modell geplant wird oder keinerlei DiGA-Modell in Aussicht steht. Außerdem klären wir die Frage: Kommt ein standardisiertes Modell in Europa für die EU-DiGA?
3. Als Schweizer Hersteller auf den EU-Markt kommen
Wenn Sie als Schweizer Hersteller ein Produkt auf den EU-Markt bringen wollen, müssen wir zunächst unterscheiden, ob Sie bereits ein Medizinprodukt haben, oder dessen Entwicklung erst bevorsteht. Beide Fälle beleuchten wir in den nachfolgenden Kapiteln.
WICHTIG: Als Schweizer Hersteller ohne Sitz in der EU benötigen Sie in jedem Fall einen Bevollmächtigten/Repräsentanten, sowie einen Importeur mit Sitz in der EU. Zudem müssen Sie Ihr Medizinprodukt in der EUDAMED Datenbank anmelden.
3.1 Sie haben noch kein Medizinprodukt
Bei der Entwicklung von Medizinprodukten für die EU gilt in erster Linie die MDR.
Da die MDR in der Schweiz anerkannt ist und die MepV an vielen Stellen darauf verweist, sind die Anforderung an die Produktentwicklung zum größten Teil identisch. Dazu zählt zum Beispiel der Aufbau eines Qualitätsmanagementsystems und die Erstellung einer technischen Dokumentation. Die genauen Anforderungen haben wir in anderen Blogartikeln bereits zusammengefasst:
- Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten (MDR)
- Leitfaden: Ist Ihre App ein Medizinprodukt?
Die Frage, ob Sie zuerst den Schweizer- (unter MepV) oder den EU-Markt (unter MDR) betreten möchten, sollten Sie zu Beginn klären. Beides hat gewisse Vor- und Nachteile, welche Sie für Ihr Unternehmen abwägen müssen. Dazu müssen Sie unter anderem herausfinden, welcher Markt für Sie interessanter ist und inwiefern Sie Ihre technische Dokumentation offenlegen möchten (z.B. an einen EU-Bevollmächtigten).
Falls Sie bereits ein Medizinprodukt entwickelt und zugelassen haben, lesen Sie im nächsten Kapitel weiter.
3.2 Sie haben bereits ein Medizinprodukt nach MepV
Wenn Sie bereits ein Medizinprodukt konform mit der MepV entwickelt haben, haben Sie für das Produkt auch ein Konformitätsbewertungsverfahren durchgeführt. Bevor Sie das Produkt nun auf den EU-Markt bringen können, müssen Sie prüfen, ob das durchgeführte Verfahren auch für die EU zulässig ist. Sofern eine bezeichnete Stelle in die Konformitätsbewertung des Medizinprodukts involviert war, stellen Sie sicher, dass diese auch als benannte Stelle nach MDR anerkannt ist. In vielen Fällen ist dies nämlich nicht der Fall und Sie müssen ein weiteres Konformitätsbewertungsverfahren gemäß MDR durchführen.
Außerdem benötigen Sie einen Bevollmächtigten und einen Importeur mit Sitz in der EU und müssen Ihr Produkt in EUDAMED anmelden. Mehr dazu erfahren Sie in den folgenden Kapiteln.
3.2.1 Konformitätsbewertungsverfahren nach MDR
Um die CE-Kennzeichnung auf einem Medizinprodukt anbringen zu können, müssen Hersteller ein sogenanntes Konformitätsbewertungsverfahren durchführen. Die MDR bietet drei verschiedene Bewertungsverfahren, welche deckungsgleich mit den Verfahren der MepV sind. Bei Software-Produkten handelt es sich dabei meist um eine Bewertung auf Basis des Qualitätsmanagementsystems und der technischen Dokumentation. Zu diesem Thema finden Sie weitere Informationen in unserem Artikel: Zulassung & Zertifizierung von Software-Medizinprodukten (MDR)
3.2.2 Bevollmächtigter & Importeur in der EU
Genauso wie EU-Hersteller in der Schweiz, benötigen auch Schweizer Hersteller in der EU einen Bevollmächtigten (auch “Repräsentant”), um ihre Produkte auf dem EU-Markt vertreiben zu können.
Der Bevollmächtigte in der EU hat dieselben Pflichten, wie sein Äquivalent in der Schweiz (siehe weiter oben) – mit einer Ausnahme:
Der EU-Repräsentant muss über eine Kopie der technischen Dokumentation und der EU-Konformitätserklärung verfügen und diese auf Anfrage den zuständigen Behörden bereitstellen können.
Genauso wie in der Schweiz, benötigen Hersteller auch innerhalb der EU einen Importeur, welcher das Produkt in die EU importiert und auf dem Markt verfügbar macht.
Sie sind Medizinprodukt-Hersteller mit Sitz in der Schweiz? Dann sprechen Sie uns an – wir übernehmen gerne die Rolle des Bevollmächtigten und des Importeurs und stellen ein reibungsloses Inverkehrbringen Ihrer Produkte in der EU sicher.
3.3.3 Anmeldung in EUDAMED
Alle Wirtschaftsakteure (Hersteller, Bevollmächtigter und Importeur) müssen in EUDAMED registriert sein. Diese Anmeldung erfolgt über das entsprechende Online-Portal, nach welcher Sie eine SRN (eindeutige Kennung) erhalten.
Auch Medizinprodukte selbst müssen in der EUDAMED Datenbank registriert werden. Alles über die Zertifizierung und Zulassung von Medizinprodukten unter MDR erfahren Sie in unserem Artikel: Zulassung & Zertifizierung von Software-Medizinprodukten (MDR)
4. Fazit
Da aktuell kein Abkommen mehr zwischen der Schweiz und der EU besteht, wird die Schweiz im Sinne der MDR als Drittland behandelt. Das bedeutet, dass Medizinprodukt-Hersteller einen Bevollmächtigten/Repräsentanten in der EU brauchen, um ein Produkt dort in Verkehr zu bringen. Andersherum verhält es sich genauso, jedoch werden Medizinprodukte mit einer CE-Kennzeichnung in der Schweiz einseitig anerkannt, sodass für EU-Hersteller keine zusätzlichen Zertifizierungspflichten bestehen.
Schweizer Hersteller hingegen müssen häufig ein weiteres Konformitätsbewertungsverfahren (unter Einbezug einer benannten Stelle) durchführen, um Ihre Medizinprodukte in der EU anbieten zu können.
Mit Sitz in Deutschland übernimmt QuickBird Medical gerne die Rolle des Bevollmächtigten und des Importeurs innerhalb der EU. Sprechen Sie uns an, wenn Sie Unterstützung bei dem Inverkehrbringen oder der Entwicklung von MDR-Medizinprodukten benötigen. Wir helfen Ihnen gerne weiter.

