

Gibt es noch Software-Medizinprodukte der Risikoklasse I nach MDR? Werden wirklich reihenweise Softwareprodukte der Risikoklasse I von Aufsichtsbehörden aus dem Verkehr gezogen? In welches Bundesland muss ich gehen, damit ich mit meiner Klasse I Software auf dem Markt bleiben darf?
Da in letzter Zeit immer mehr Gerüchte kursieren, dass eine Zulassung von Software-Medizinprodukten unter Risikoklasse I nicht mehr möglich ist, möchten wir mit diesem Artikel etwas Licht ins Dunkel bringen.
Dieser Artikel richtet sich vor allem an Hersteller von Medizinprodukt-Software, von denen keine oder nur geringe Risiken ausgehen. Das umfasst beispielsweise Trainingsapps, die von Patienten autonom genutzt werden und die keine Schäden am Patienten verursachen können.
Update – Juni 2025: Das Guidance-Dokument „MDCG 2019-11: Guidance on Qualification and Classification of Software […]“ liegt seit Juni 2025 in der überarbeiteten Revision 1 vor. In diesem Artikel sind sämtliche Anpassungen berücksichtigt. Eine detaillierte Übersicht der konkreten Änderungen finden Sie auch hier.
Update – Dezember 2025: Die EU-Kommission hat im Dezember 2025 einen Änderungsentwurf zur MDR veröffentlicht, der unter anderem eine Anpassung der Regel 11 zur Klassifizierung von Software-Medizinprodukten vorsieht. Auf die möglichen Auswirkungen für Risikoklasse-I-Software gehen wir im Kapitel 3 dieses Artikels ein.
Inhaltsverzeichnis
- 1. Risikoklassen der MDR
- 2. Update der offiziellen MDCG-Guidance (Revision 1) – Juni 2025
- 3. Änderungsentwurf der MDR & Implikationen für Klasse I-Software – Dezember 2025
- 4. Umgang mit Unsicherheit bei der Risikoklasse
- 5. Warum wir Risikoklasse I in der EU dringend brauchen
- 6. Fazit
1. Risikoklassen für Medizinprodukte der MDR
Da wir die Risikoklassen der MDR bereits in unserem Software-Klassifizierungs-Guide umfassend beschrieben haben, möchten wir es an dieser Stelle kurz halten:
- Die MDR unterscheidet bei Software im Kern die Risikoklassen I, IIa, IIb und III
- Entscheidend für die Bestimmung der Risikoklasse eines Produkts ist seine Zweckbestimmung
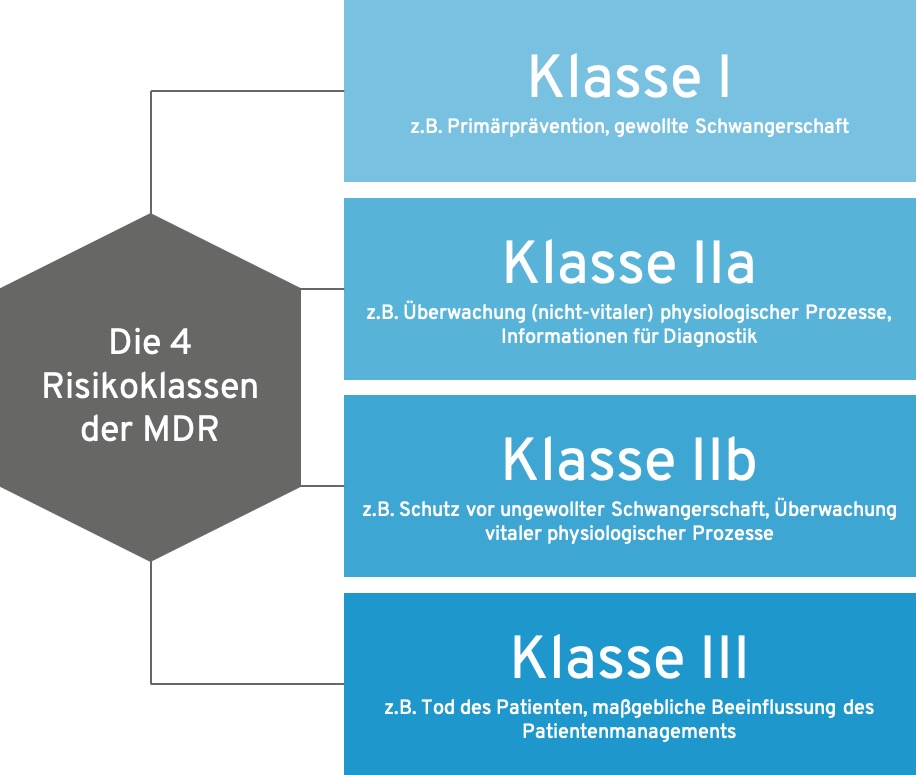
Die 4 MDR-Risikoklassen im Überblick
Falls Sie mehr über die Risikoklassifizierung von Software-Medizinprodukten erfahren möchten, helfen Ihnen folgende Leitfäden von uns:
- Qualifizierung von Software-Medizinprodukten – Leitfaden: Ist Ihre App ein Medizinprodukt?
- Klassifizierung von Software-Medizinprodukten – Leitfaden: In welche Risikoklasse fallen Sie?
Die Zulassung von Produkten der Risikoklasse I ist im Vergleich zu höherklassigen Produkten sehr viel ressourcenschonender und schneller möglich. Daher stellen sich viele Hersteller die Frage, ob es sich bei ihrem Produkt um ein Produkt der Klasse I oder IIa handelt.
Warum das so ist, erfahren Sie im nächsten Kapitel.
1.1 Risikoklasse I oder IIa – Warum ist das wichtig?
Warum ist die Unterscheidung zwischen Klasse I und IIa überhaupt so wichtig? Und warum wird so viel darüber diskutiert?
Produkte der Risikoklasse I unterscheiden sich maßgeblich von höherklassigen Produkten.
Klasse I-Produkte …
- benötigen bei Marktzulassung kein Zertifikat einer benannten Stelle,
- können ohne Verzögerung (verursacht durch einen Audit) auf den Markt gehen und müssen keine Kosten für eine benannte Stelle tragen,
- werden lediglich unangekündigten Prüfungen durch die jeweilige Aufsichtsbehörde des Bundeslands unterzogen. Diese sind deutlich weniger aufwendig als die Zertifizierung bei einer benannten Stelle (Risikoklasse IIa und höher) und nicht kostenpflichtig.
Um das Ganze noch besser zu veranschaulichen, möchten wir hier einige Erfahrungswerte aufzeigen. Ein Klasse IIa Produkt bedeutet im Vergleich zu Klasse I:
- Verzögerung der Marktzulassung um 6–18 Monate (Dauer des Konformitätsbewertungsverfahrens einer benannten Stelle und Ausstellung des CE-Zertifikats)
- Kosten bis zu 6-stelligen Eurobeträgen für das Konformitätsbewertungsverfahren und die Zertifizierung des QMS
- Pflicht der Anzeige signifikanter Änderungen und weitere Prüfungen der benannten Stelle bei Produktänderungen nach der Zulassung
Wie lange es bis zum erfolgreichen Abschluss eines Konformitätsbewertungsverfahrens dauert, ist höchst individuell und hängt von verschiedenen Faktoren ab. In jedem Fall handelt es sich dabei um eine weitere Unbekannte, die man in der Planung seines Produkts nur schwer abbilden kann.
Die Unterscheidung zwischen Risikoklasse I und IIa entscheidet daher in vielen Fällen wirklich über die Existenz von Produkten und ganzen Unternehmen.
Deshalb ist Klasse I vor allem für Start-ups interessant, die auf eine schnelle Marktzulassung angewiesen sind und nur begrenzte Ressourcen haben.
Was genau bei einem Risikoklasse IIa-Produkt im Vergleich zu Klasse I auf Sie zukommt, erfahren Sie in einem gesonderten Fachartikel von uns: MDR Risikoklasse I vs. IIa: Unterschiede für Software-Medizinprodukte
1.2 MDR – Regel 11 – Klare Grenze zwischen Klasse I und IIa?
Die Antwort, ob Ihre Software oder App in die Risikoklasse I fallen kann, findet sich in der Medical Device Regulation (MDR) – Regel 11.
Zitat der MDR 6.3. Regel 11 (verkürzt):
Software, die dazu bestimmt ist, Informationen zu liefern, die zu Entscheidungen für diagnostische oder therapeutische Zwecke herangezogen werden, gehört zur Klasse IIa,
[…]
Sämtliche andere Software wird der Klasse I zugeordnet.
In diesem Bezug fragen sich viele Hersteller:
-
- Was sind Informationen, die zu Entscheidungen für diagnostische und therapeutische Zwecke herangezogen werden?
- Wer kann solche Informationen überhaupt “heranziehen”, um diagnostische oder therapeutische Entscheidungen zu treffen?
- Kann eine App, die einem Patienten nur Trainingsmodule und Wissen vermittelt, überhaupt die Therapie oder Diagnostik beeinflussen?
- Ist die Empfehlung, eine bestimmte Übung zu machen, schon eine Therapieentscheidung?
Überraschung: Es gibt verschiedene Möglichkeiten, diese Regel 11 zu interpretieren. Denn das BfArM, benannte Stellen und Hersteller sind sich hier nicht einig. Es existiert viel Interpretationsspielraum, weshalb es immer wieder zu Diskussionen und Gerüchten kommt. Daher möchten wir hier die 2 wichtigsten Interpretationen dieser Regel 11 beschreiben.
1.2.1 Interpretation 1: Jede Software liefert Informationen, die die Therapie oder Diagnostik beeinflussen
Nach dieser Interpretation liefert auch eine App für Patienten Informationen für Entscheidungen zur Therapie oder Diagnose von Krankheiten. Der Patient würde sich ja selbst therapieren bzw. diagnostizieren und somit greife Satz 1 aus Regel 11.
Mit dieser Interpretation gibt es streng genommen keine Klasse I Standalone-Software, da Software definitionsgemäß immer einen Output (Informationen) auf Basis eines Inputs erzeugt. Eine Ausnahme wäre beispielsweise eine App, die jedem Nutzer den exakt gleichen Inhalt anzeigt, ohne irgendeine Individualisierung. Doch hier stellt sich die Frage, ob es sich dann überhaupt um ein Medizinprodukt handelt (siehe auch unser Leitfaden „Ist Ihre App ein Medizinprodukt?“).
1.2.2 Interpretation 2: Nur medizinisches Fachpersonal kann therapeutische oder diagnostische Entscheidungen treffen
Das ist die herstellerfreundliche Interpretation der Regel 11. Demnach würde eine Trainingsapp, die lediglich von Patienten verwendet wird, in Risikoklasse I fallen. Die Begründung: Ein Laie ist nicht dazu in der Lage, therapeutische oder diagnostische Entscheidungen zu treffen. Satz 1 aus Regel 11 greift daher nicht – Informationen werden nicht für Entscheidungen für diagnostische oder therapeutische Zwecke herangezogen.
Hinweis: Anders sieht es aber bei Produkten aus, die auch medizinisches Fachpersonal in die Nutzung einbeziehen und unterstützende Informationen liefern (z. B. über ein Web Dashboard). Dies wurde beispielsweise vom Hanseatische Oberlandesgericht im Fall von Dermanostic entschieden. In diesem Fall hat ein Mitbewerber ein Gerichtsverfahren gegen Dermanostic eingeleitet. Das Gericht entschied dann, dass es sich bei Dermanostic um ein Produkt der Klasse IIa handelt und nicht um eines der Klasse I. Mehr dazu lesen Sie hier.
1.2.3 Wessen Interpretation zählt nun?
Wir sehen, dass verschiedene Stakeholder unterschiedliche Auffassungen vertreten:
-
- BfArM
- Landesaufsichtsbehörde
- Benannte Stelle
- Hersteller
- Gericht
Wenn Sie sich dazu entscheiden, den Weg zum Risikoklasse I Produkt zu gehen, ist vor allem Ihre eigene Meinung (Hersteller), aber natürlich auch die Meinung Ihrer Landesaufsichtsbehörde relevant. Diese Behörde ist nämlich in erster Linie verantwortlich für die Überwachung der Hersteller in Ihrer Region.
Auch die Entscheidung von Gerichten zählt natürlich, sofern es zu einem Rechtstreit (z.B. zwischen Ihnen und einem Konkurrenten) kommt – siehe Dermanostic .
Die Einschätzung benannter Stellen ist vor allem dann wichtig, wenn Sie Risikoklasse IIa oder höher anstreben. Bei Produkten der Risikoklasse I sind diese nämlich gar nicht eingebunden.
Das BfArM hat vor allem indirekten Einfluss auf die Risikoklassifizierung. Da es sich dabei um die Bundesbehörde handelt, kann das BfArM auch Einfluss auf die Landesaufsichtsbehörden ausüben und z.B. Empfehlungen herausgeben, denen Landesbehörden ggf. folgen.
1.3 Schlussfolgerung – geht Klasse I noch?
Ja, eine Zulassung von Software-Medizinprodukten der Risikoklasse I ist noch möglich. Allerdings ist das stark von Ihrer Landesaufsichtsbehörde abhängig. Unter Umständen kann es sich lohnen, den Sitz Ihres Unternehmens in Bundesländer zu verlagern, deren Aufsichtsbehörden „Klasse I-freundlich“ sind.
Alternativ gibt es ein weiteres spannendes Modell, das wir anbieten. Sie können uns als externen Inverkehrbringer beauftragen, Ihr Medizinprodukt auf den Markt zu bringen. Wir bieten diesen Service für Software-basierte Medizinprodukte und DiGA an: Mehr Informationen
Sprechen Sie uns gerne an, wenn Sie einen externen Inverkehrbringer für Ihre App oder andere Standalone-Software benötigen.
2. Update der offiziellen MDCG-Guidance (Revision 1) – Juni 2025
Die offizielle MDCG-Guidance zur Qualifizierung und Klassifizierung von Medical Device Software (MDSW) wurde im Juni 2025 in Revision 1 aktualisiert. Das Update wurde lange erwartet und erzeugte große Unsicherheit in der Digital Health-Szene. Die zentrale Frage im Raum: Bedeutet die neue Version das Ende von Risikoklasse I-Software?
Das ist glücklicherweise nicht der Fall. Es hat sich tatsächlich wenig in Bezug auf die (teilweise unkonkreten) Empfehlungen zur Risikoklasse I vs. IIa getan. Wir haben hier eine detaillierte Übersicht zu allen Änderungen erstellt: Alle Änderungen der MDCG 2019-11 Rev.1
3. Änderungsentwurf der MDR & Implikationen für Klasse I-Software – Dezember 2025
Am 16. Dezember 2025 hat die EU-Kommission einen Änderungsentwurf zur Medical Device Regulation (MDR) veröffentlicht. Ein zentraler Bestandteil dieses Entwurfs ist eine grundlegende Überarbeitung der Regel 11, welche wir in der aktuell gültigen Fassung oben erklärt haben.
Auf den ersten Blick wirkt der neue Wortlaut für Hersteller vielversprechend: Die Logik der Klassifizierung wird scheinbar umgedreht, sodass Software zunächst in Klasse I fällt, sofern keine höherklassifizierenden Kriterien greifen. Bei genauerer Analyse zeigt sich jedoch, dass diese Hoffnung trügerisch sein könnte. Je nach Auslegung würde der Entwurf dazu führen, dass ein Großteil medizinischer Software mindestens in Klasse IIa eingeordnet werden würde.
Wir haben den Änderungsentwurf, die neue Regel 11 sowie deren mögliche Interpretationen detailliert analysiert und die konkreten Auswirkungen auf Klasse-I-Software in einem separaten Fachartikel aufgearbeitet: Zum Fachartikel
Wichtig ist dabei: Der veröffentlichte Text ist ein früher Entwurf im europäischen Gesetzgebungsverfahren. Erfahrungsgemäß wird der Wortlaut in den kommenden Jahren weiter angepasst. Es ist daher unwahrscheinlich, dass dieser Entwurf für die Regel 11 unverändert in den finalen MDR-Text übernommen wird.
4. Umgang mit Unsicherheit bei der Risikoklasse
Wie kann ich sicher sein, dass meine Risikoklasse richtig ist? Eine absolute Rechtssicherheit gibt es bei diesem Thema eigentlich nie. Grundsätzlich ist eine höhere Klassifizierung immer “sicherer”, jedoch auch sehr viel aufwendiger und teurer.
Da sich ein solches Verfahren aber viele Firmen nicht leisten wollen oder können, möchten wir hier ein paar Möglichkeiten aufzeigen, die Sie als Hersteller eines Klasse-I-Produkts haben.
- Lesen Sie die MDCG Guidance Dokumente (v.a. MDCG 2019-11).
- Sehen Sie sich ähnliche Produkte auf dem Markt an, die unter MDR zugelassen sind. Dabei sollten Sie vor allem Produkte von Herstellern berücksichtigen, für die Ihre auch Ihre Aufsichtsbehörde verantwortlich ist.
- Holen Sie Erfahrungswerte von Experten ein. Es gibt viele Berater, die in die Zulassung vieler Klasse I-Medizinprodukte involviert waren. Diese Erfahrungen sind in vielen Fällen auf ihr Produkt übertragbar. Sprechen Sie uns gerne an, wenn Sie dabei Unterstützung benötigen.
- Holen Sie ein juristisches Gutachten ein. Ein solches wird von einem Anwalt ausgestellt und kann Ihre Argumentation vor einer Aufsichtsbehörde, aber auch vor Gericht untermauern. Es schafft zusätzliche Überzeugungskraft. Beachten Sie aber, dass ein solches Gutachten keine absolute Rechtssicherheit geben kann. Sprechen Sie uns an, wenn Sie an einem Gutachten interessiert sind. Wir vermitteln Sie gerne an einen Anwalt, der sich mit der Thematik auskennt.
4.1 Was passiert, wenn ich mein Produkt unter Risikoklasse I anmelde, obwohl ich mir unsicher bin?
Grundsätzlich können Sie das natürlich tun. Es liegt in der Verantwortung des Herstellers, die Klassifizierung seiner Produkte ordnungsgemäß durchzuführen.
ABER seien Sie sich bewusst, dass dies ein unternehmerisches Risiko darstellen kann. Einige dieser Risiken sind:
| Szenario | Gefahr | Schaden |
| Das Produkt birgt wirklich Risiken in der Anwendung | Ein Patient könnte sich verletzen | Rechtliche Konsequenzen, Reputationsschaden |
| Ein Mitbewerber leitet Gerichtsverfahren gegen Sie ein | Das Gericht entscheidet, dass Sie Ihr Produkt höher klassifizieren müssen | Produkt muss vom Markt genommen oder höher klassifiziert werden |
| Eine Aufsichtsbehörde prüft Ihr Produkt und widerspricht der Risikoklassifizierung | Die Aufsichtsbehörde ist anderer Meinung in Bezug auf die Risikoklasse Ihres Produkts | Produkt muss vom Markt genommen oder höher klassifiziert werden |
5. Warum wir Risikoklasse I in der EU dringend brauchen
Es gibt unzählige innovative Software-Produkte mit medizinischem Anwendungsfall, von denen kaum Risiken für Patienten ausgehen (z.B. Trainingsapps zur Reduktion von Ängsten). Der Zugang zu solchen Produkten wurde in den letzten Jahren (z.B. durch die Einführung von DiGA) enorm gefördert und tausende Patienten nutzen solche Produkte täglich effektiv zur Verbesserung ihres Gesundheitszustands. Das entlastet nicht nur Patienten, sondern das gesamte Gesundheitssystem.
Der Aufwand für eine Zulassung unter Risikoklasse IIa steht aus unserer Sicht in keinem Verhältnis zu den meist sehr geringen Risiken, die von Patienten-fokussierten Laienprodukten ausgehen.
Solche Produkte kommen zudem oft aus dem Startup-Umfeld, welches meist mit limitierten Ressourcen auskommen muss. Durch eine Überregulierung (und hart gesagt – das Unmöglich-Machen von Klasse I Software) erstickt man solche Innovationen im Keim. Kleine Unternehmen können es sich schlichtweg nicht leisten, ein Konformitätsbewertungsverfahren mit einer benannten Stelle zu durchlaufen – weder zeitlich, noch finanziell.
Das ist nicht nur schlecht für den Wirtschaftsstandort Deutschland, da Hersteller ihre Standorte in andere Länder verlagern, sondern am Ende auch eine politische Verhinderung einer besseren Patientenversorgung.
Wir benötigen daher dringend weiterhin die Möglichkeit, Software der Risikoklasse I zu entwickeln und auch zuzulassen.
6. Fazit zur Risikoklasse I nach MDR
Nach all den Gerüchten und Diskussionen stand die Frage im Raum: „Gibt es noch Standalone-Software-Medizinprodukte der Risikoklasse I?“
Die Situation ist zwar nach viel Recherche und zahlreichen Gesprächen in unserem Umfeld weiterhin nicht final geklärt, allerdings lassen sich einige Erkenntnisse gewinnen:
-
- Aufsichtsbehörden, benannte Stellen und Hersteller interpretieren die MDR unterschiedlich, weshalb bis heute kein klarer Konsens in Bezug auf die Risikoklasse I bei Software herrscht.
- Das offizielle Update der MDCG-Guidance hat wenig in Bezug auf die bisherige Unklarheit zwischen Risikoklasse I und IIa geändert. Das ist eine sehr gute Nachricht. Denn das bedeutet unterm Strich: Es wurde zumindest nicht gewollt, dass Risikoklasse I z. B. für DiGA und Übungs-Apps explizit ausgeschlossen wird. Es ist davon auszugehen, dass Risikoklasse I auch in Zukunft von den meisten Aufsichtsbehörden in Bezug auf risikoarme Übungs-Apps akzeptiert wird. Ob das BfArM sich hierzu irgendwann eine andere Meinung bildet, bleibt abzuwarten.
Wir von QuickBird Medical sind der Meinung, dass es unbedingt Softwareprodukte der Risikoklasse I geben muss. Die Risiken, die von solchen Produkten ausgehen, sind verschwindend klein, haben aber in der Masse einen riesigen Einfluss auf eine bessere Patientenversorgung. Eine höhere Risikoklasse senkt die Attraktivität erheblich, solche Produkte zu entwickeln und zuzulassen. Die damit einhergehenden Aufwände sind für viele Unternehmen nicht zu stemmen oder schlicht zu riskant. Unser Gesundheitssystem – und vor allem Patienten – profitiert aber enorm von Produkten der Risikoklasse I.
Abonnieren Sie unseren Newsletter, um zur Risikoklassifizierung auf dem neuesten Stand zu bleiben.
Sprechen Sie uns auch gerne an, wenn Sie die Realisierung eines Software-Medizinprodukts planen. Wir entwickeln regulierte Apps, Web-Anwendungen und andere medizinische Software auf Auftragsbasis für andere Unternehmen.

