

Die EU reguliert mit der KI-Verordnung (EU AI Act) nun auch den Einsatz von künstlicher Intelligenz (KI). Dies betrifft auch Hersteller von Software-Medizinprodukten, die KI in ihre MDR-Medizinprodukte integrieren („Medical Device Artificial Intelligence“ bzw. MDAI). Der AI Act soll dafür sorgen, dass KI-Systeme so gestaltet sind, dass die Sicherheit und die Grundrechte von Personen geschützt werden.
- Aber ist die KI-Verordnung für mein Medizinprodukt überhaupt relevant?
- Welche zusätzlichen Anforderungen müssen speziell Medizinprodukt-Hersteller umsetzen?
- Und wie schaffe ich es, diese in mein bestehendes Qualitätsmanagementsystem zu integrieren?
Diese und weitere Fragen beantworten wir in diesem umfangreichen Leitfaden zum EU-AI Act. Wir fokussieren uns hierbei komplett auf die Auswirkungen der KI-Verordnung auf MDR-Medizinprodukte.
Inhalte des Leitfadens zur KI-Verordnung
- 1. Was ist der AI Act? (Stand 2026)
- 2. Ist der AI Act für mein Produkt relevant?
- 3. Die 3 Produktkategorien des AI Act
- 4. Anforderungen an Hochrisiko-KI-Systeme
- 5. Wie beeinflusst der AI Act die Zulassung meines Medizinprodukts?
- 6. Ab wann gilt der AI Act? Umsetzungs-Timeline
- 7. Fazit
1. Was ist der EU AI Act?
Der AI Act ist eine neue Verordnung, welche die Entwicklung und den Einsatz von künstlicher Intelligenz (KI, oder AI für Artificial Intelligence) in der EU regeln soll. Sie ist nicht auf Medizinprodukte beschränkt, sondern adressiert grundsätzlich jegliche Produkte, die unter die Definition eines KI-Systems fallen (siehe nächstes Kapitel “Ist der AI Act für mein Produkt relevant?”). Das übergeordnete Ziel der KI-Verordnung ist es, die Sicherheit und Grundrechte von Personen zu wahren. Die gesamte Verordnung finden Sie auf der Webseite der Webseite der Europäischen Union.
2. Ist der AI Act für mein Produkt relevant?
Der AI Act definiert KI-Systeme folgendermaßen:
Für die Zwecke dieser Verordnung bezeichnet der Ausdruck „KI-System“ ein maschinengestütztes System, das für einen in unterschiedlichem Grade autonomen Betrieb ausgelegt ist und das nach seiner Betriebsaufnahme anpassungsfähig sein kann und das aus den erhaltenen Eingaben für explizite oder implizite Ziele ableitet, wie Ausgaben wie etwa Vorhersagen, Inhalte, Empfehlungen oder Entscheidungen erstellt werden, die physische oder virtuelle Umgebungen beeinflussen können;
Zusammengefasst ist ein KI-System demnach eine Software, die aus den erhaltenen Inputs ableitet, wie ein bestimmter Output erzeugt werden kann. Zudem kann das System (aber muss nicht) autonom arbeiten und laufend weiterlernen. Unterm Strich ist damit zwar noch nicht 100% klar, welche Systeme davon genau exkludiert sind, aber da die meisten Produkte mit KI unter Machine Learning fallen, sind sie sehr eindeutig von dieser Definition erfasst.
Die MDCG hat ein Dokument veröffentlicht, welches das Zusammenspiel zwischen der MDR und dem AI Act verdeutlicht: MDCG 2025-6. Dieses Dokument ist speziell für Hersteller von Software-Medizinprodukten interessant. Dort werden Produkte, die sowohl der MDR, als auch dem AI Act unterliegen, als „Medical Device Artificial Intelligence (MDAI)“ definiert.
3. Die 3 Produktkategorien des AI Act
Gelten für alle KI-Medizinprodukte (MDAI) dieselben Anforderungen? Die kurze Antwort lautet: Nein. Ähnlich wie die MDR unterteilt auch der EU AI Act Produkte in verschiedene Klassen:
- Verbotene KI Praktiken
- Hochrisiko-KI-Systeme
- Andere KI-Systeme (mit geringem Risiko)
In welche Klasse fällt mein KI- bzw. Software-Medizinprodukt? Und was hat das für mich als Hersteller zu bedeuten? 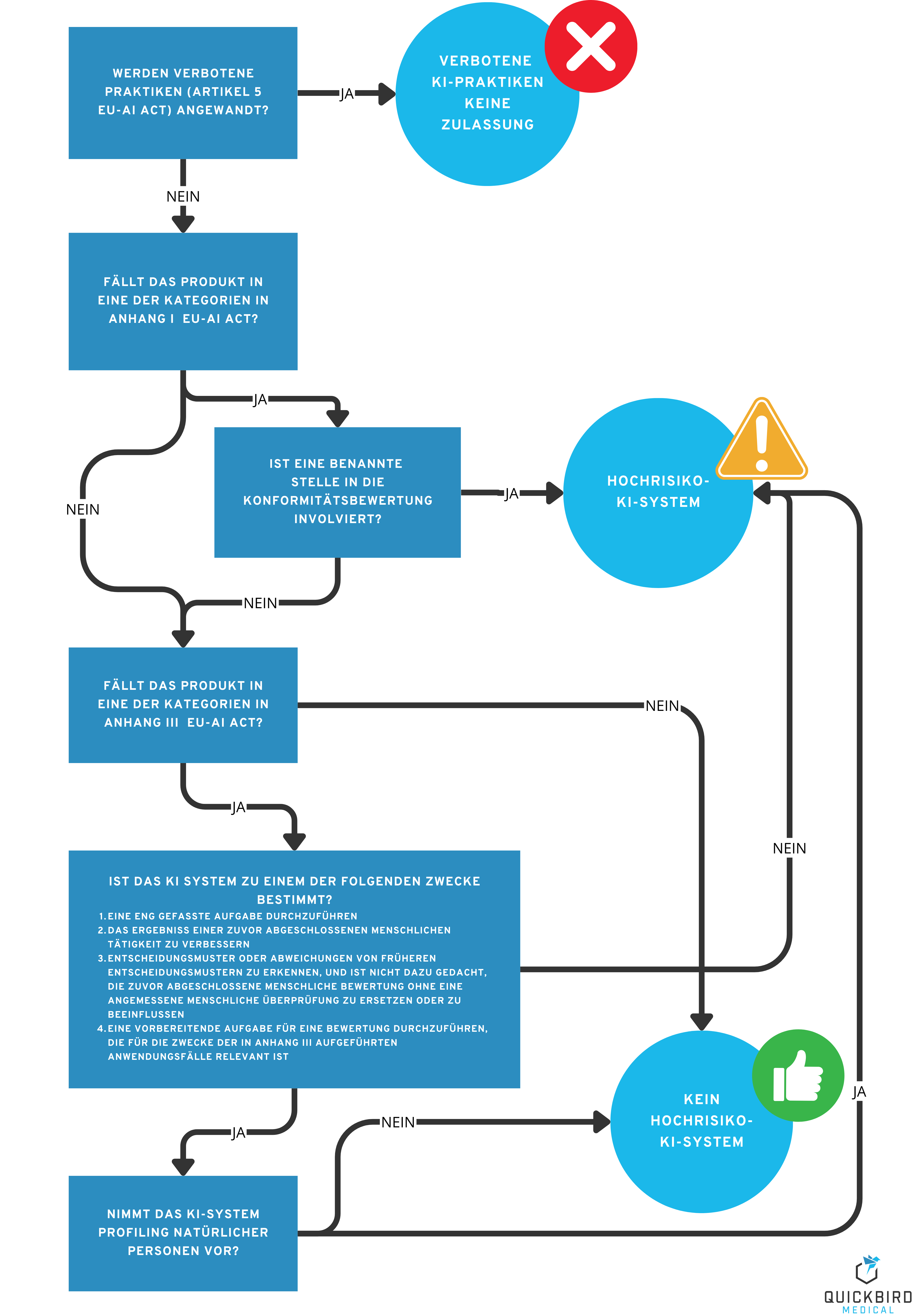
Entscheidungsbaum: In welche KI-Risikoklasse fällt mein KI- bzw Software-Mizinprodukt (MDAI)?
3.1 Produktkategorie 1: Verbotene KI-Praktiken
Auf das Wesentliche reduziert lässt sich sagen, dass vor allem solche Praktiken verboten sind, die zum Ziel haben, Personen zu schaden – und zwar durch unterschwellige Beeinflussung oder das Ausnutzen bestimmter Schwächen oder Schutzbedürftigkeiten (z.B. Behinderungen, Alter). Artikel 5 der KI-Verordnung listet noch eine Reihe weiterer verbotener Praktiken, die aber in der Regel für Medizinprodukte nicht relevant sind.
Fällt mein Medizinprodukt in diese Kategorie? Das Ziel, Personen zu schaden, widerspricht üblicherweise den Zielen eines Medizinprodukts, weshalb Sie grundsätzlich davon ausgehen können, dass Ihre KI nicht in die Kategorie der “verbotenen Praktiken” fällt. Dennoch sollten Sie sich die Kriterien in Artikel 5 genau ansehen, um sicherzugehen.
Welche Anforderungen muss ich bei „Verbotenen KI-Praktiken“ beachten? Das Produkt darf nicht auf den Markt gebracht werden.
3.2 Produktkategorie 2: Hochrisiko-KI-Systeme
In diese Kategorie fallen Produkte, die ein hohes Risiko für die Gesundheit und Sicherheit, oder die Grundrechte natürlicher Personen darstellen.
Fällt mein Medizinprodukt in diese Kategorie? Vor allem bei den Begriffen “Gesundheit” und “Sicherheit” liegt der Gedanke an “Medizinprodukte” nicht mehr fern, zumal auch die MDR einen starken Fokus auf die Sicherheit, der von ihr geregelten Produkte legt.
Gemäß MDCG 2025-6 ist ein Medizinprodukt dann ein Hochrisiko-KI-System, wenn folgende Kriterien erfüllt sind:
- Das KI-System ist selbst ein Medizinprodukt oder eine Sicherheitskomponente davon
- Es ist eine benannte Stelle in die Konformitätsbewertung nach MDR eingebunden
Wenn in das Konformitätsbewertungsverfahren Ihres Produkts eine benannte Stelle involviert ist (z.B., weil es in Risikoklasse IIa oder höher fällt), ist Ihr Medizinprodukt automatisch ein Hochrisiko-KI-System gemäß KI-Verordnung (siehe Entscheidungsbaum in der Abbildung oben). Die benannte Stelle hat dann nicht nur die Anforderungen der MDR, sondern auch jene des AI Act im Zuge der Konformitätsbewertung zu prüfen. Wenn aber keine benannte Stelle bei der Zulassung Ihres Produkts involviert ist (z.B., weil das Produkt in Risikoklasse I nach MDR fällt, oder es sich gar nicht um ein Medizinprodukt handelt), müssen Sie vor allem Anhang III des AI Acts prüfen. Dieser enthält eine Liste von Systemen, die als Hochrisiko-KI-Systeme gelten. Die meisten Medizinprodukte werden sich aber nicht in dieser Liste finden lassen, weshalb wir davon ausgehen, dass viele Medizinprodukte der Risikoklasse I nach MDR keine Hochrisiko-KI-Systeme sind.
Diese Einschätzung bestätigt auch die MDCG 2025-6, in welcher die untenstehende Tabelle aufgeführt ist:
 Einordnung von Hochrisiko-KI-Systemen nach MDR
Einordnung von Hochrisiko-KI-Systemen nach MDR
Welche Anforderungen muss ich beachten? Wenn Ihr Produkt in die Kategorie der Hochrisiko-KI-Systeme fällt, hat die KI-Verordnung möglicherweise starke Auswirkungen auf Ihre Organisation und Ihr Medizinprodukt. Die Inhalte des EU AI Acts definieren nämlich speziell Anforderungen an die Hersteller solcher Systeme und die Systeme selbst. Einen Überblick über die Anforderungen geben wir Ihnen weiter unten im Kapitel “Anforderungen an Hochrisiko-KI-Systeme”.
3.3 Produktkategorie 3: Andere KI-Systeme (mit geringem Risiko)
Alle KI-Produkte, die weder den verbotenen KI-Praktiken noch den Hochrisiko-KI-Systemen zugeordnet werden können, fallen in diese Kategorie. Es handelt sich dabei grob gesagt um Systeme, von denen keine nennenswerten Risiken für die Sicherheit und Grundrechte von Personen ausgehen. Fällt mein Medizinprodukt in diese Kategorie? Wenn Ihr Produkt die folgenden Kriterien erfüllt, fällt es vermutlich in diese Kategorie:
- Es handelt sich um ein Produkt der Risikoklasse I nach MDR (es ist keine benannte Stelle in die Konformitätsbewertung involviert)
- Es wurde ausgeschlossen, dass es sich um ein Hochrisiko-KI-System handelt
- Es wurde ausgeschlossen, dass es sich um verbotene KI-Praktiken handelt
Welche Anforderungen muss ich beachten? Im Vergleich zu Hochrisiko-KI-Systeme kommen Produkte in dieser Kategorie wirklich gut weg. Im Grunde findet sich im gesamten AI Act nur eine Anforderung an solche Systeme: Transparenzpflichten. Und diese gelten auch nur für solche Systeme, die mit Menschen interagieren, mediale Inhalte erzeugen, oder zur Emotionserkennung dienen. Grob gesagt müssen Hersteller solcher Produkte sicherstellen, dass Nutzer wissen, dass sie es mit KI zu tun haben, und dass es sich bei den erzeugten Inhalten um keine echten Bilder, Videos, etc. handelt (z.B. Deepfake). KI-Systeme dieser Kategorie müssen in keiner gesonderten Datenbank angemeldet werden und sie bedürfen keiner Konformitätserklärung. Wenn Ihr Medizinprodukt also in diese Kategorie fällt, sind die Aufwände für die Konformität mit dem AI Act für Sie vermutlich überschaubar.
3.4 Zusatzkategorie: KI-Modelle mit allgemeinem Verwendungszweck
Unabhängig von der Risiko-Einstufung können bestimmte KI-Modelle als „KI-Modell mit allgemeinem Verwendungszweck“ (Englisch: „General-purpose AI model“) gelten. Das sind Modelle, die für verschiedene Zwecke verwendet werden können.
Da der Zweck von Medizinprodukten aber meist sehr genau definiert ist, wird diese Kategorie in der Praxis nur für wenige Hersteller relevant sein. Der Vollständigkeit halber möchten wir sie an dieser Stelle aber trotzdem nennen.
Falls Sie ein KI-Modell entwickeln, das über die Anwendung in einem konkreten Produkt hinausgeht, sollten Sie sich mit dieser Kategorie näher beschäftigen.
Die genauen Anforderungen an solche Modelle sind in Kapitel V des AI Act definiert.
4. KI-Verordnung – Anforderungen an Hochrisiko-KI-Systeme und deren Hersteller
Die genauen Anforderungen an Hochrisiko-KI-Systeme und ihre Hersteller sind in Kapitel III Abschnitt 2 und Abschnitt 3 des AI Act spezifiziert. Wir gehen an dieser Stelle davon aus, dass Sie bereits die Anforderungen der MDR erfüllen. Somit gehen wir hier vor allem auf die Punkte ein, die über die Anforderungen der MDR hinausgehen. Erfahren Sie mehr über die Anforderungen der MDR an KI-Medizinprodukte in diesem Artikel: Zulassung & Zertifizierung von Software-Medizinprodukten (MDR)
Die KI-Verordnung definiert die Pflichten der Anbieter von Hochrisiko-KI-Systemen wie folgt (Artikel 16). Sie müssen …
a) sicherstellen, dass ihre Hochrisiko-KI-Systeme die in Abschnitt 2 festgelegten Anforderungen erfüllen;
b) auf dem Hochrisiko-KI-System oder, falls dies nicht möglich ist, auf seiner Verpackung oder in der beigefügten Dokumentation ihren Namen, ihren eingetragenen Handelsnamen bzw. ihre eingetragene Handelsmarke und ihre Kontaktanschrift angeben;
c) über ein Qualitätsmanagementsystem verfügen, das Artikel 17 entspricht;
d) die in Artikel 18 genannte Dokumentation aufbewahren;
e) die von ihren Hochrisiko-KI-Systemen automatisch erzeugten Protokolle gemäß Artikel 19 aufbewahren, wenn diese ihrer Kontrolle unterliegen;
f) sicherstellen, dass das Hochrisiko-KI-System dem betreffenden Konformitätsbewertungsverfahren gemäß Artikel 43 unterzogen wird, bevor es in Verkehr gebracht oder in Betrieb genommen wird;
g) eine EU-Konformitätserklärung gemäß Artikel 47 ausstellen;
h) die CE-Kennzeichnung an das Hochrisiko-KI-System oder, falls dies nicht möglich ist, auf seiner Verpackung oder in der beigefügten Dokumentation anbringen, um Konformität mit dieser Verordnung gemäß Artikel 48 anzuzeigen;
i) den in Artikel 49 Absatz 1 genannten Registrierungspflichten nachkommen;
j) die erforderlichen Korrekturmaßnahmen ergreifen und die gemäß Artikel 20 erforderlichen Informationen bereitstellen;
k) auf begründete Anfrage einer zuständigen nationalen Behörde nachweisen, dass das Hochrisiko-KI-System die Anforderungen in Abschnitt 2 erfüllt;
l) sicherstellen, dass das Hochrisiko-KI-System die Barrierefreiheitsanforderungen gemäß den Richtlinien (EU) 2016/2102 und (EU) 2019/882 erfüllt.
Keine Sorge, wir lassen Sie mit dieser Liste nicht allein. Im Folgenden wollen wir deshalb beleuchten, was das für Sie als Hersteller eines KI-Medizinprodukts bedeutet. Wir gehen dabei auf die wichtigsten Punkte ein, die zu einer Anpassung Ihres Managementsystems und/oder des Medizinprodukts führen.
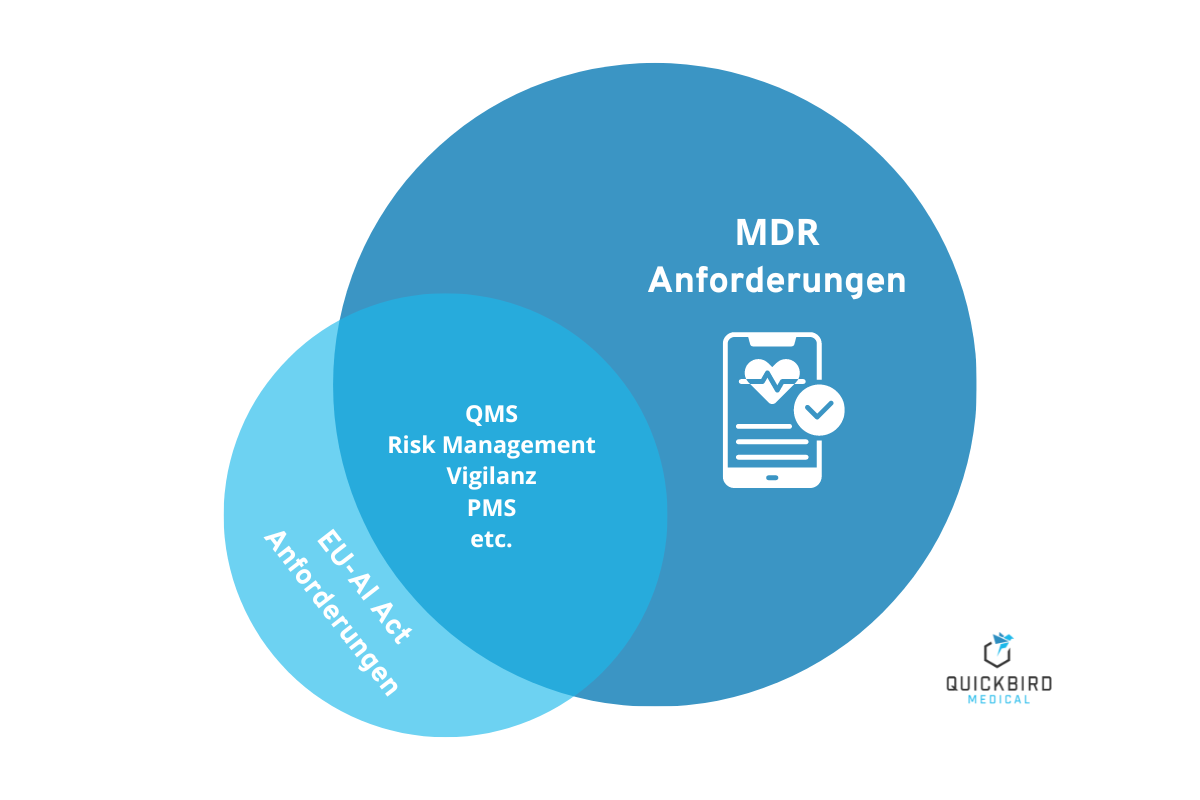
Die Anforderungen des EU-AI Act und der MDR überschneiden sich in zahlreichen Punkten.
4.1 AI Act – Anforderungen an Ihr Qualitätsmanagementsystem
Ein Medizinprodukthersteller wird von der MDR bereits dazu verpflichtet, ein Qualitätsmanagementsystem zu etablieren. In den meisten Fällen wird dabei unter anderem die ISO 13485 und – wenn es um Software geht – die IEC 62304 umgesetzt. Auf den ersten Blick sieht es so aus, als hätten Sie damit auch die Anforderung des AI Acts erfüllt. Auf den zweiten Blick wird aber deutlich, dass dies nur teilweise stimmt. Aber keine Angst, der Großteil Ihres Systems kann bleiben, wie er ist. Sie müssen lediglich ein paar Anpassungen und Erweiterungen vornehmen. Die Anforderungen aus Artikel 17 des EU AI Acts finden Sie in der untenstehenden Tabelle aufgelistet. Zusätzlich zu den genauen Anforderungen der Verordnung haben wir die konkreten Maßnahmen für Sie definiert, die Sie umsetzen müssen, um die entsprechende Konformität Ihres Qualitätsmanagementsystems herzustellen. Das Qualitätsmanagementsystem muss mindestens die folgenden Aspekte umfassen:
(Hinweis: Unsere Kommentare sind nicht als absolute Wahrheiten zu verstehen, sondern sollen lediglich etwaige Gemeinsamkeiten und Unterschiede zwischen dem AI Act und Ihrem bestehenden System verdeutlichen. Prüfen Sie daher für jeden Punkt, inwiefern Sie Maßnahmen ergreifen müssen.)
| Wortlaut aus dem AI Act | Kommentar zur Umsetzung |
|---|---|
| a) ein Konzept zur Einhaltung der Regulierungsvorschriften, was die Einhaltung der Konformitätsbewertungsverfahren und der Verfahren für das Management von Änderungen an dem Hochrisiko-KI-System miteinschließt; | Da Sie als Medizinprodukt-Hersteller bereits ein Konzept zur Einhaltung der MDR, DSGVO und anderer regulatorischer Vorgaben haben, sollten Sie dieses einfach um den AI Act erweitern. |
| b) Techniken, Verfahren und systematische Maßnahmen für den Entwurf, die Entwurfskontrolle und die Entwurfsprüfung des Hochrisiko-KI-Systems; | Dieser Punkt könnte bereits durch die ISO 13485 und IEC 62304 umgesetzt sein. |
| c) Techniken, Verfahren und systematische Maßnahmen für die Entwicklung, Qualitätskontrolle und Qualitätssicherung des Hochrisiko-KI-Systems; | Dieser Punkt sollte durch die ISO 13485 und IEC 62304 bereits umgesetzt sein. |
| d) Untersuchungs-, Test- und Validierungsverfahren, die vor, während und nach der Entwicklung des Hochrisiko-KI-Systems durchzuführen sind, und die Häufigkeit der Durchführung; | Auch dieses Thema findet sich in der ISO 13485 wieder und als Medizinprodukt-Hersteller verfügen Sie bereits über entsprechende Prozesse. Prüfen Sie aber, ob Ihre Verfahren für das KI-System angemessen sind. |
| e) die technischen Spezifikationen und Normen, die anzuwenden sind und, falls die einschlägigen Normen nicht vollständig angewandt werden oder sie nicht alle relevanten Anforderungen gemäß Abschnitt 2 abdecken, die Mittel, mit denen gewährleistet werden soll, dass das Hochrisiko-KI-System diese Anforderungen erfüllt; | Harmonisierte Normen zur Umsetzung des AI Act werden aber im laufe der Zeit von der Europäischen Kommission festgelegt (Stand vom 20. Januar 2026). Dies wird vermutlich ähnlich aussehen, wie unter der MDR, deren harmonisierte Normen in einem Durchführungsbeschluss veröffentlicht sind und laufend erweitert werden. |
| f) Systeme und Verfahren für das Datenmanagement, einschließlich Datengewinnung, Datenerhebung, Datenanalyse, Datenkennzeichnung, Datenspeicherung, Datenfilterung, Datenauswertung, Datenaggregation, Vorratsdatenspeicherung und sonstiger Vorgänge in Bezug auf die Daten, die im Vorfeld und für die Zwecke des Inverkehrbringens oder der Inbetriebnahme von Hochrisiko-KI-Systemen durchgeführt werden; | Diese Verfahren müssen vermutlich neu etabliert werden, da sie nicht explizit von der MDR, oder den damit assoziierten Normen gefordert werden. Mehr dazu finden Sie weiter unten im Kapitel “Daten und Daten-Governance”. |
| g) das in Artikel 9 genannte Risikomanagementsystem; | Dieser Punkt sollte zum Großteil bereits durch die MDR (bzw. ISO 14971) abgedeckt sein. Prüfen Sie, inwieweit Ihr Risikomanagement die Anforderungen des AI Act schon erfüllt (siehe folgende Kapitel). |
| h) die Einrichtung, Anwendung und Aufrechterhaltung eines Systems zur Beobachtung nach dem Inverkehrbringen gemäß Artikel 72; | Dieser Punkt ist zum Großteil bereits durch die MDR (Post-Market Surveillance) abgedeckt. Prüfen Sie, inwieweit Ihr System zur Beobachtung nach dem Inverkehrbringen die Anforderungen des AI Act schon erfüllt (siehe folgende Kapitel). |
| i) Verfahren zur Meldung eines schwerwiegenden Vorfalls gemäß Artikel 73; | Dieser Punkt ist zum Großteil bereits durch die MDR (Vigilanz) abgedeckt. Für Medizinprodukte gibt es hier sogar eine Sonderregelung. Dadurch, dass Medizinprodukt-Hersteller bereits über ein solches System verfügen, fordert die KI-Verordnung lediglich, dass die neuen Anforderungen in dieses System integriert werden. Inwiefern diese überhaupt zu Änderungen an Ihrem System führen, müssen Sie prüfen. Es kann gut sein, dass hier wenig, bis gar kein Aufwand entsteht. |
| j) die Handhabung der Kommunikation mit zuständigen nationalen Behörden, anderen einschlägigen Behörden, auch Behörden, die den Zugang zu Daten gewähren oder erleichtern, notifizierten Stellen, anderen Akteuren, Kunden oder sonstigen interessierten Kreisen; | Die Kommunikation mit verschiedenen Stakeholdern müssen Sie durch die ISO 13485 bereits regeln. Erweitern Sie diese Liste also einfach um etwaige neue Behörden, oder Akteure, um Konformität mit diesem Punkt zu erlangen. Dies könnte beispielsweise dann relevant werden, wenn Sie Daten eines KI-Reallabors nutzen wollen. |
| k) Systeme und Verfahren für die Aufzeichnung sämtlicher einschlägigen Dokumentation und Informationen; | Diesen Punkt sollten Sie durch Ihr Qualitätsmanagementsystem schon umgesetzt haben. Hier geht es darum, Prozesse zu etablieren, die die notwendigen Outputs (z.B. technische Dokumentation) generieren. |
| l) Ressourcenmanagement, einschließlich Maßnahmen im Hinblick auf die Versorgungssicherheit; | Diesen Punkt sollten Sie durch Ihr Qualitätsmanagementsystem nach ISO 13485 bereits umgesetzt haben. |
| m) einen Rechenschaftsrahmen, der die Verantwortlichkeiten der Leitung und des sonstigen Personals in Bezug auf alle in diesem Absatz aufgeführten Aspekte regelt. | Dies Verantwortlichkeit können Sie auf Ihre PRRC nach MDR übertragen, oder eine neue Rolle dafür definieren. |
4.1.1 Risikomanagement
Als Medizinprodukt-Hersteller verfügen Sie bereits über ein Risikomanagementsystem und sind ggf. von verschiedenen Gesetzen zu Risikomanagement-Aktivitäten verpflichtet (z.B. MDR, DSGVO). Die Anforderungen an das Risikomanagementsystem sind aber in MDR und AI Act nicht deckungsgleich! Die wohl fundamentalste Unterschied ist, dass Sie nicht nur Risiken in Bezug auf die Sicherheit, sondern auch auf die Grundrechte von Personen berücksichtigen müssen. Teilweise kann dies durch die Einhaltung anderer Verordnungen (z.B. DSGVO) zwar umgesetzt sein, neben der körperlichen Unversehrtheit und dem Schutz personenbezogener Daten können aber auch andere Rechte durch ein KI-System verletzt werden. Werfen Sie daher auf jeden Fall einen Blick in die Grundrechte-Charta der Europäischen Union. Zudem müssen Hochrisiko-KI-Systeme mit geeigneten Verfahren regelmäßig getestet werden (dies gilt vermutlich vor allem für kontinuierlich weiterlernende Systeme, die unter der MDR nur in Ausnahmefällen zulassungsfähig sind). Dieses Testen ist auch erforderlich, um die geeignetsten Risikomanagementmaßnahmen zu ermitteln. Die ermittelten Risiken sind dabei so weit wie „technisch möglich“ und bis zu einem „vertretbar beurteilbaren“ Grad gesenkt werden.
4.1.2 System zur Beobachtung nach dem Inverkehrbringen
Durch die MDR müssen Sie bereits die Post-Market Surveillance Ihres Medizinprodukts sicherstellen. Darauf wird in der KI-Verordnung auch explizit eingegangen. Wenn Sie nämlich bereits ein solches System nach MDR etabliert haben, müssen Sie prüfen, ob dieses auch die folgende Anforderung erfüllt: Das System muss es Ihnen ermöglichen, die Konformität des Produkts fortlaufend bewerten zu können.
4.1.3 System zur Meldung schwerwiegender Vorfälle und Fehlfunktionen
Durch Ihr bereits etabliertes Vigilanz-System nach MDR gibt es in diesem Bereich wenig umzusetzen. Der AI Act hat hier sogar eine Sonderregel für Medizinprodukte. Dadurch, dass bereits eine Verpflichtung zur Meldung sicherheitsrelevanter Vorfälle besteht, müssen Sie Ihr System lediglich um die Meldung von “Verstößen gegen die Bestimmungen des Unionsrechts zum Schutz der Grundrechte” erweitern.
4.1.4 Daten und Daten-Governance
Hier werden Sie als Hersteller dazu verpflichtet, geeignete Prozesse zu etablieren, welche Folgendes regeln:
- Konzeptionelle Entscheidungen (z.B. Wahl des Modelltyps, Festlegung relevanter Features)
- Datenerfassung (z.B. Datenquellen)
- Datenaufbereitungsvorgänge (z.B. Labelling, Bereinigung, Aggregierung)
- Aufstellung relevanter Annahmen (z.B. Beziehung zwischen Features, potenzielle Einflussfaktoren)
- Vorherige Bewertung der Verfügbarkeit, Menge und Eignung der benötigten Datensätze
- Untersuchung im Hinblick auf mögliche Verzerrungen/Bias und geeignete Maßnahmen zu derer Erkennung, Verhinderung und Abschwächung
- Ermittlung möglicher Datenlücken, Mängel und Maßnahmen, diese zu beheben
Grundsätzlich stellt dieses Kapitel auch konkrete Anforderungen an Ihre Trainings-, Validierungs- und Testdaten. Diese Anforderungen sollten Sie direkt in Ihre Prozesse integrieren. Tipp: Berücksichtigen Sie bei der Erstellung dieser Prozesse auch die Anforderungen an die technische Dokumentation in Anhang IV und Artikel 11 der KI-Verordnung. Dadurch stellen Sie sicher, dass Ihre Verfahren auch die notwendigen Outputs generieren.
4.1.5 Aufbewahrungspflichten
Wenn Sie die Aufbewahrungspflichten der MDR einhalten, erfüllen Sie vermutlich auch jene des AI Acts. Dort ist definiert, dass Sie Ihre Dokumentation für einen Zeitraum von 10 Jahren nach dem Inverkehrbringen des KI-Systems aufbewahren müssen. Zu dieser Dokumentation zählt laut Artikel 18 der KI-Verordnung die technische Dokumentation, das Qualitätsmanagementsystem, von benannten Stellen freigegebene Änderungen und ausgestellte Dokumente, sowie die Konformitätserklärung. Vom Produkt automatisch erzeugte Protokolle müssen 6 Monate lang aufbewahrt werden.
4.2 AI Act – Anforderungen an die technische Dokumentation
„Technische Dokumentation“ ist ein Begriff, der Ihnen sicherlich bereits aus der MDR bekannt ist. Eine technische Dokumentation müssen Sie für Ihr Medizinprodukt ohnehin erstellen. Tatsächlich gibt es auch hier viele Parallelen und Sie werden die meisten Punkte bereits durch die Einhaltung der MDR umgesetzt haben. Darunter zählen beispielsweise die Produktbeschreibung, eine Konformitätserklärung und die Methoden, die bei der Produktentwicklung angewandt wurden. Dies gilt aber nicht in jedem Fall! Besonders für Software-Systeme der Software-Sicherheitsklasse A nach IEC 62304 könnten sich in Bezug auf die technische Dokumentation einige Besonderheiten ergeben. Die KI-Verordnung verpflichtet Sie z.B. dazu, eine Systemarchitektur und die Definition von Schnittstellen zu anderen Systemen zu dokumentieren. Dies geht über die Anforderungen der IEC 62304 für Software-Systeme der Sicherheitsklasse A hinaus. Tipp: Pauschal kann man also nicht sagen, in welchem Umfang Ihre technische Dokumentation erweitert werden muss. Gehen Sie den Anhang IV des AI Acts also am besten durch und prüfen Sie für jeden Punkt, inwiefern Sie diesen durch Ihre aktuelle Dokumentation bereits umgesetzt haben. Alle noch fehlenden Informationen erstellen Sie im Anschluss idealerweise im Zuge der neu definierten bzw. angepassten Prozesse.
4.3 Anforderungen an das Produkt
4.3.1 Protokollierung und Logs
Hochrisiko-KI-Systeme sind zur Protokollierung verpflichtet. Es handelt sich hier also um eine klare technische Vorgabe. Diese Protokollierung muss, wie so oft im “der Zweckbestimmung angemessenen Maße” stattfinden. Was genau das heißt, wird in Artikel 12 der KI-Verordnung definiert. Demnach geht es vor allem um die Aufzeichnung von Ereignissen, die für folgendes relevant sind: a) die Ermittlung von Situationen, die dazu führen können, dass das Hochrisiko-KI-System ein Risiko im Sinne des Artikels 79 Absatz 1 birgt oder dass es zu einer wesentlichen Änderung kommt, b) die Erleichterung der Beobachtung nach dem Inverkehrbringen gemäß Artikel 72 und c) die Überwachung des Betriebs der Hochrisiko-KI-Systeme gemäß Artikel 26 Absatz 5. Im Kern geht es also darum, Situationen, die mit Risiken und Schäden verbunden sind möglichst gut zu überwachen und rückverfolgen zu können. Bei der Umsetzung dieser Protokollierung müssen sich Hersteller an anerkannte Standards und Spezifikationen halten (z.B. Anforderungen des BSI).
4.3.2 Transparenz und Bereitstellung von Informationen für die Nutzer
Wenn Sie noch keine Gebrauchsanweisungen für Ihr Medizinprodukt haben, brauchen Sie spätestens jetzt eine. Sollten Sie bereits eine Gebrauchsanweisung haben, müssen Sie diese vermutlich überarbeiten. Auch wenn es einige Anforderungen gibt, die auch schon von der MDR gestellt werden, fordert der AI Act nun auch Informationen zu z.B. Cyber Security und Risiken für die Grundrechte. Hier müssen auch Genauigkeitsgrade und relevante Genauigkeitskennzahlen des Systems angegeben werden. Prüfen Sie also für Ihre aktuelle Gebrauchsanweisung, inwiefern Sie die Pflichten des AI Acts bereits erfüllen und machen Sie anschließend die notwendigen Anpassungen.
4.3.3 Menschliche Aufsicht
Die Pflicht zur menschlichen Aufsicht ist eine verpflichtende Risikokontrollmaßnahme für Hochrisiko-KI-Systeme. Das System muss so konzipiert und entwickelt werden, dass es von einem Menschen wirksam beaufsichtigt werden kann. Hier lassen sich einige Parallelen zu den Transparenzanforderungen erkennen. Im Grunde muss es einem Nutzer möglich sein, die Vorgänge im Produkt, sowie dessen Ausgaben zu verstehen. Zudem muss er in der Lage sein, das System abzuschalten oder in den Betrieb einzugreifen. Die Möglichkeit zur Aufsicht kann dabei entweder direkt in die Software eingebaut werden, oder auch an den Nutzer abgegeben werden. Grob zusammengefasst muss es schlichtweg möglich sein, Anomalien oder Fehlverhalten zu erkennen und das System im Zweifelsfall stoppen zu können.
4.3.4 Genauigkeit, Robustheit und Cybersicherheit
Hochrisiko-KI-Systeme müssen widerstandsfähig gegenüber Fehlern, Störungen oder Unstimmigkeiten sein, die innerhalb des Systems oder der Umgebung, in der das System betrieben wird, insbesondere wegen seiner Interaktion mit natürlichen Personen oder anderen Systemen auftreten können. Die Robustheit von Hochrisiko-KI-Systemen kann durch technische Redundanz erreicht werden, was auch Sicherungs- oder Störungssicherheitspläne umfassen kann. Hochrisiko-KI-Systeme, die nach dem Inverkehrbringen (oder der Inbetriebnahme) kontinuierlich weiterlernen, sind so zu entwickeln, dass auf möglicherweise verzerrte Ergebnisse mit geeigneten Risikominderungsmaßnahmen eingegangen wird. Hochrisiko-KI-Systeme müssen widerstandsfähig gegen Versuche unbefugter Dritter sein, ihre Verwendung oder Leistung durch Ausnutzung von Systemschwachstellen zu verändern. Die technischen Lösungen zur Gewährleistung der Cybersicherheit von Hochrisiko-KI-Systemen müssen den jeweiligen Umständen und Risiken angemessen sein. Die technischen Lösungen für den Umgang mit KI-spezifischen Schwachstellen umfassen gegebenenfalls Maßnahmen zur Verhütung und Kontrolle von Angriffen, mit denen versucht wird, den Trainingsdatensatz zu manipulieren („Datenvergiftung“), von Eingabedaten, die das Modell zu Fehlern verleiten sollen („feindliche Beispiele“), oder von Modellmängeln.
4.3.5 CE-Kennzeichnung
Eine CE-Kennzeichnung hat Ihr Medizinprodukt bereits, wenn Sie es konform im Rahmen der MDR zugelassen haben. Sie müssen nur noch die entsprechende Verordnung referenzieren, damit Nutzer nachvollziehen können, dass Ihr Produkt im Einklang mit den beiden Verordnungen (MDR und AI Act) entwickelt worden ist.
4.3.6 Registrierung des Produkts
Ähnlich wie EUDAMED für Medizinprodukte, wird es in Zukunft auch eine EU-Datenbank für Hochrisiko-KI-Systeme geben, in welcher Ihr Produkt registriert werden muss. Zum aktuellen Zeitpunkt ist eine solche aber noch nicht öffentlich zugänglich (Stand 20.01.2026).
5. Wie beeinflusst der AI Act die Zulassung meines Medizinprodukts?
Die Risikoklasse Ihres Medizinprodukts nach MDR hat Einfluss auf die Anwendbarkeit des AI Acts. Wie Sie die Risikoklasse nach MDR bestimmen, erfahren Sie in unserem Leitfaden zur Klassifizierung von Software-Medizinprodukten.
5.1 Risikoklasse I
Bei der Zulassung von Produkten der Risikoklasse I nach MDR ändert sich durch den AI Act nichts (bis auf die Anforderungen aus Kapitel 3.3) – solange das Produkt kein Hochrisiko-KI-System darstellt. Sollte dies aber der Fall sein, brauchen Sie für die Konformitätsbewertung in Zukunft eine benannte Stelle, welche die Konformität mit der KI-Verordnung prüft. Behalten Sie dies unbedingt im Hinterkopf, wenn Sie die Umsetzung eines Produkts der Risikoklasse I planen, da es den Markteintritt maßgeblich verzögern kann.
Achtung: Es gelten Ausnahmen für Produkte der Risikoklasse Im, Is und Ir, bei denen eine benannte Stelle in die Konformitätsbewertung involviert ist. Diese sind bei reinen Software-Produkten allerdings eher selten.
5.2 Risikoklasse IIa oder höher
Produkte der Risikoklasse IIa oder höher sind im Grunde automatisch auch Hochrisiko-KI-Systeme gemäß dem AI Act. Somit sind einige Zusatzanforderungen umzusetzen, die weiter oben im Artikel bereits ausgeführt wurden. Bei der Zulassung (insbesondere der Konformitätsbewertung) müssen Sie in Zukunft unbedingt darauf achten, dass Ihre benannte Stelle auch unter der KI-Verordnung akkreditiert ist, oder eine solche Akkreditierung anstrebt. Diese muss im Zuge der Konformitätsbewertung dann nicht nur die Konformität mit der MDR, sondern auch mit dem AI Act prüfen.
Die Konformitätsbewertung wird dann voraussichtlich aber nicht für beide Verordnungen separat, sondern im Zuge einer integrierten Prüfung durchgeführt.
Nur, wenn das Produkt mit beiden Verordnungen konform ist, darf es das CE-Symbol tragen. Setzen Sie sich also schnellstmöglich mit Ihrer benannten Stelle in Verbindung und klären Sie, ob eine solche Akkreditierung für die KI-Verordnung geplant ist.
Die MDCG hat im Juni 2025 in diesem Zusammenhang ein Dokument veröffentlich, welches das Zusammenspiel zwischen MDR und AI Act beschreibt: MDCG 2025-6
6. Ab wann gilt der AI Act? Umsetzungs-Timeline
Der EU AI Act tritt nicht sofort komplett in Kraft, sondern erst über die Zeit. Die aktuelle Timeline sieht vor, dass der Großteil der Verordnung bis 2. August 2026 in Kraft getreten ist und sie ab dem 2. August 2027 dann vollständig gültig ist.
Das geht aus der Timeline der Europäischen Kommission hervor:
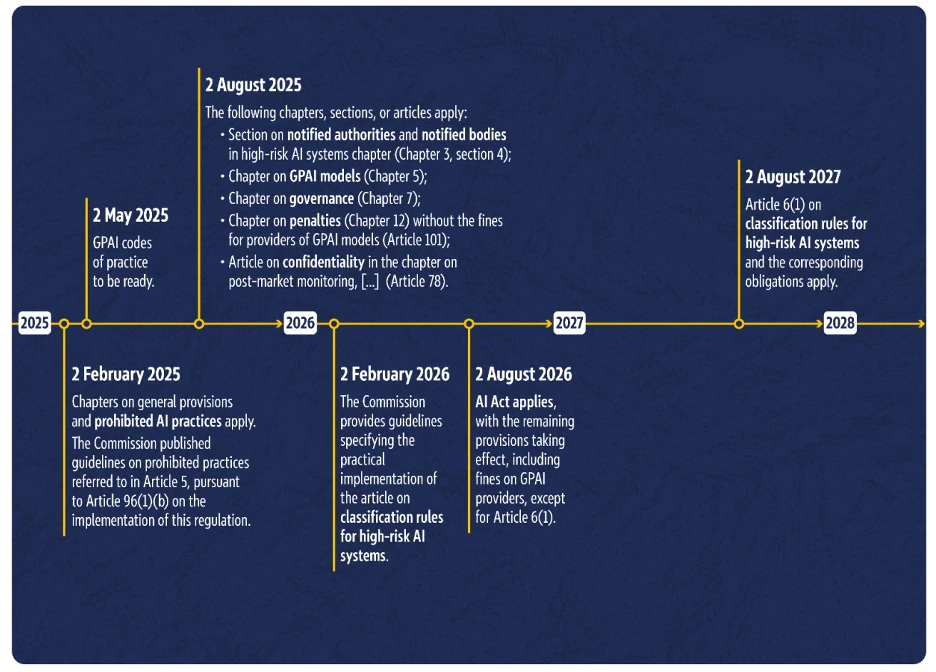
Umsetzungs-Zeitplan für den AI Act der Europäischen Kommission (Quelle: AI Act implementation timeline)
Zusammengefasst bedeutet dies für Hersteller von KI- bzw. Software-Medizinprodukten (MDAI):
- Viele grundsätzliche Regeln des AI Act sind bereits heute gültig (z.B. Verbotene KI-Praktiken)
- Ab August 2026 gilt fast der komplette AI Act, allerdings mit einer wichtigen Ausnahme: Artikel 6(1). Dieser beschreibt die Einstufungsregeln für Hochrisiko-KI-Systeme.
- Hersteller von Hochrisiko-KI-Systemen haben somit bis August 2027 Zeit, alle Anforderungen an ihr Produkt und an sich selbst als Hersteller umzusetzen (außer, es ist ein System, welches in Anhang III genannt ist). Denn erst ab diesem Zeitpunkt gilt die Einstufung als Hochrisiko-KI-System.
7. Fazit
Der AI Act unterscheidet sich von der MDR zu allererst durch seine Zielsetzung. Während die MDR v.a. darauf abzielt, die Sicherheit und Funktionstauglichkeit von Medizinprodukten sicherzustellen, schließt die KI-Verordnung auch den Schutz der Grundrechte von Personen mit ein. Grundsätzlich lässt sich sagen, dass vor allem auf MDR-Medizinprodukte der Risikoklasse IIa oder höher viele neue Anforderungen zukommen. Diese werden nämlich automatisch der Kategorie “Hochrisiko-KI-Systeme” gemäß des AI Acts zugeordnet. Doch auch Medizinprodukte der Risikoklasse I können im Einzelfall in diese Kategorie fallen. Für Produkte, die keine Hochrisiko-KI-Systeme darstellen und nicht zu den “verbotenen Praktiken” zählen, bringt die KI-Verordnung wenig Änderungen. Hersteller von Hochrisiko-KI-Medizinprodukten müssen einer Reihe von Stellen Anpassungen vornehmen, um die Anforderungen des AI Act einzuhalten. Neben den Anforderungen des AI Acts, müssen Sie natürlich auch die Anforderungen der MDR beachten, welche in Bezug auf künstliche Intelligenz relevant sind.
Benötigen Sie Unterstützung bei der Entwicklung eines medizinischen KI-Systems? Wir entwickeln medizinische KI-Systeme für Unternehmen im Gesundheitswesen auf Auftragsbasis.
Unsere KI-Services: – Technischer Review von medizinischen KI-Systemen – Entwicklung von neuen Machine Learning-Modellen im Gesundheitsbereich – Individuelle Beratung bezüglich regulatorischer Compliance für Ihr geplantes KI-basiertes Produkt
Wir helfen Ihnen, auch die umfangreichen Anforderungen der MDR und des AI Act umzusetzen und ein erfolgreiches Produkt zu entwickeln.

