

Viele In-vitro-Diagnostika bestehen heute nicht mehr nur aus physischen Testkits, sondern aus komplexer Software, die Analyseergebnisse berechnet, interpretiert oder visualisiert. Damit fällt immer mehr Software in den Anwendungsbereich der In-vitro-Diagnostika-Verordnung (In Vitro Diagnostic Regulation, abgekürzt IVDR).
Diese Verordnung regelt Produkte, die Proben außerhalb des Körpers untersuchen, zum Beispiel Blut-, Urin- oder Gewebeproben. Der Begriff „in vitro“ bedeutet wörtlich „im Glas“ und beschreibt genau diese Untersuchungen außerhalb des menschlichen Körpers.
Dieser Artikel richtet sich an (angehende) Hersteller von IVD-Produkten, welche entweder komplett oder teilweise aus Software bestehen. Wir erklären Ihnen, wann Software unter die In Vitro Diagnostic Regulation (IVDR) fällt, wie die Qualifizierung funktioniert und nach welchen Kriterien die Risikoklassifizierung erfolgt.
Aus unserer praktischen Erfahrung in Kundenprojekten zur Umsetzung von IVDR-Software (und sogar mobilen IVD-Apps) können wir sagen: Die Einordnung ist nicht immer ganz einfach. Wir brechen das Thema für Sie daher herunter und teilen es in praxisnahe Unteraspekte auf. So erhalten Sie eine klare Grundlage, um die Qualifizierung und Klassifizierung Ihres Produkts vorzunehmen.
Inhaltsverzeichnis
- 1. Qualifizierung: Fällt Ihre Software unter die IVDR?
- 2. Klassifizierung: Unter welche Risikoklasse fällt Ihre Software?
- 3. Wer trifft die finale Entscheidung zur Qualifizierung & Klassifizierung?
- 4. Gemeinsamkeiten und Unterschiede: MDR vs. IVDR
- 5. Fazit
1. Qualifizierung: Fällt Ihre Software unter die IVDR?
Die Qualifizierung dient dazu, herauszufinden, ob Ihre Software überhaupt als In-vitro-Diagnostikum gilt und damit unter die IVDR fällt. Die Basis hierfür stellt die Zweckbestimmung (mehr dazu hier) Ihres Produkts dar.
Für die Qualifizierung sollten Sie folgende 3 Aspekte für Ihr Produkt prüfen.
1.1 Aspekt 1: Definition eines IVD nach IVDR
Zusammengefasst fällt Ihre Software in den regulierten Bereich der IVDR, wenn diese in die Definition eines „In-vitro-Diagnostikum“ nach IVDR Artikel 2(2) fällt. Der Wortlaut dieser Definition ist der Folgende:
„In-vitro-Diagnostikum“ bezeichnet ein Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibrator, Kontrollmaterial, Kit, Instrument, Apparat, Gerät, Software oder System — einzeln oder in Verbindung miteinander — vom Hersteller zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschließlich Blut- und Gewebespenden, bestimmt ist und ausschließlich oder hauptsächlich dazu dient, Informationen zu einem oder mehreren der folgenden Punkte zu liefern
a) über physiologische oder pathologische Prozesse oder Zustände,
b) über kongenitale körperliche oder geistige Beeinträchtigungen,
c) über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
d) zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
e) über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
f) zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Prüfen Sie daher genau, ob Sie die Zweckbestimmung Ihres Produkts in dieser Definition wiederfinden.
1.2 Aspekt 2: Definition eines Medizinprodukts nach MDR
Im ersten Satz der obigen Definition sieht man, dass ein IVD auch ein „Medizinprodukt“ sein muss:
„In-vitro-Diagnostikum“ bezeichnet ein Medizinprodukt, das als Reagenz […]
Die IVDR verweist für die Definition eines Medizinprodukts auf die Medical Device Regulation (MDR). Daher müssen Sie zusätzlich zur obigen Definition auch die Definition eines Medizinprodukts mit der Zweckbestimmung Ihres Produkts abgleichen.
Wie genau die Definition eines Medizinprodukts nach MDR aussieht und wie Sie entscheiden, ob Sie mit Ihrem Produkt in diese Definition fallen, erfahren Sie in unserem Leitfaden: Ist Ihre Software ein Medizinprodukt?
Wichtig: Obwohl Sie unter die Definition eines Medizinprodukts nach MDR fallen, müssen Sie für ein In-Vitro-Diagnostikum nur die IVDR einhalten. Es gilt nicht gleichzeitig die MDR und die IVDR für Ihr Produkt.
1.3 Aspekt 3: Embedded, Zubehör oder Standalone-Software?
Neben der Beschäftigung mit der IVD-Definition, ist es aber vor allem auch wichtig, zu verstehen, wie die „regulatorische Architektur“ Ihres Gesamtprodukts aussieht:
- Wird die Software als Teil Ihres Hardware-Geräts mit-zertifiziert?
- Ist die Software gesondert zu behandeln und wird demnach als eigenes IVD-Produkt zertifiziert?
- Ist die Software ein Zubehör zu dem eigentlichen IVD-Produkt (z. B. Hardware)?
Die Antworten auf diese Fragen beeinflussen stark den Zulassungsprozess Ihres Produkts und sollten daher bereits zu Beginn der Entwicklung gefunden werden.
Hierfür müssen Sie entscheiden, in welche der folgenden drei Kategorien Ihre Software-Komponente fällt:
| Kategorie | Erklärung | Auswirkungen |
| #1: Teil eines IVD-Geräts | Software ist fest mit einem IVD-Gerät verbunden und steuert dieses oder beeinflusst seine Nutzung. Sie übernimmt Funktionen, die für den Betrieb des Geräts notwendig sind. | Software und Gerät werden als ein zusammengehöriges Medizinprodukt zertifiziert. |
| #2: Zubehör für ein IVD-Gerät | Software ist selbst kein IVD, wird aber genutzt, um die Funktion eines IVD zu unterstützen. Sie ermöglicht oder verbessert die Nutzung eines anderen IVD. Siehe Definition von „Zubehör“ in der IVDR. | Zubehör wird unabhängig vom eigentlichen IVD, mit dem es verwendet wird, gesondert klassifiziert. |
| #3: Standalone-IVD-Software | Software arbeitet unabhängig von einem physischen Gerät und erzeugt unabhängig diagnostische Informationen, basierend auf Proben- oder Messdaten. | Software wird als getrenntes IVD nach IVDR zertifiziert. |
1.4 Zusammenfassung: Aspekte der Qualifizierung
Für Sie gilt es also herauszufinden:
- Ist Ihre Software Teil eines Geräts, Zubehör oder Standalone-Software?
- Fällt Ihre Software unter die Definition eines Medizinprodukts nach MDR?
- Fällt Ihre Software unter die Definition eines In-Vitro-Diagnostikums nach IVDR?
Sie sollten diese Themen zu Beginn Ihres Vorhabens durchlaufen, um die Anwendbarkeit der IVDR für Ihr Produkt zu prüfen.
1.5 Schritt-für-Schritt-Anleitung zur Qualifizierung
Die Antwort auf die oben genannten Kernfragen ist nicht immer ganz eindeutig. Daher gibt es zusätzliche Guidance-Dokumente zur IVDR (und MDR), die einen offiziellen Charakter haben.
Bezüglich der Qualifizierung von Software in Bezug auf die IVDR sollten Sie konkret einen Blick auf die „MDCG 2019-11“ werfen. Diese versucht u.a., den Entscheidungsprozess im Rahmen der IVDR von Software für Sie zu vereinfachen bzw. zu konkretisieren: Link zur MDCG Guidance
Guidance-Dokumente der MDCG sind zwar genau genommen nicht rechtlich bindend, werden aber in der Praxis auch meist von benannten Stellen herangezogen, um derartige Entscheidungen im Rahmen der Zertifizierung zu treffen.
Wir haben den Entscheidungsbaum der MDCG 2019-11 auf Deutsch übersetzt und die Formulierungen zur besseren Verständlichkeit etwas vereinfacht:
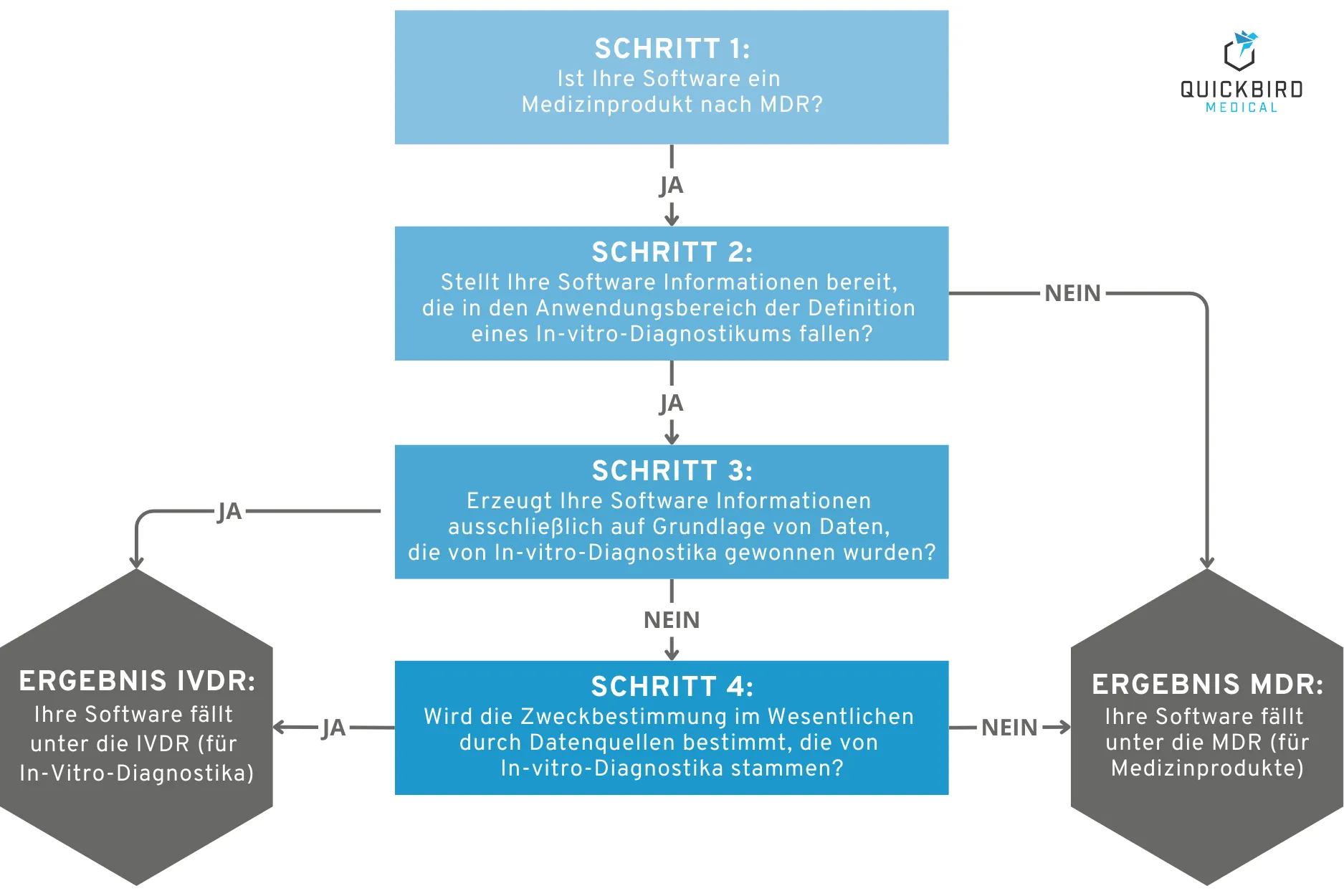
Entscheidungsbaum zur Qualifizierung: Fällt Ihre Software unter die IVDR?
Schritt 1: Ist Ihre Software ein Medizinprodukt nach MDR?
Fallen Sie mit der Zweckbestimmung Ihrer Software in die Definition eines Medizinprodukts nach MDR? Hierfür lohnt sich ein Blick auf unseren Leitfaden zu genau diesem Thema: Leitfaden: Ist Ihre Software ein Medizinprodukt?
Schritt 2: Stellt Ihre Software Informationen bereit, die in den Anwendungsbereich der Definition eines In-vitro-Diagnostikums fallen?
Konkret formuliert: Stellt Ihre Software Informationen zu einem der folgenden Zwecke zur Verfügung? (siehe Definition eines IVD in der IVDR):
a) über physiologische oder pathologische Prozesse oder Zustände,
b) über kongenitale körperliche oder geistige Beeinträchtigungen,
c) über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
d) zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
e) über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
f) zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Schritt 3: Erzeugt Ihre Software Informationen ausschließlich auf Grundlage von Daten, die von In-vitro-Diagnostika gewonnen wurden?
Sind Sie sicher, dass die Daten nicht auch aus anderen Quellen wie z. B. einem Medizinprodukt (kein IVD) stammen? Nur dann können Sie diese Frage mit „Ja“ beantworten.
Schritt 4: Wird die Zweckbestimmung im Wesentlichen durch Datenquellen bestimmt, die von In-vitro-Diagnostika stammen?
Wenn es auch Informationen gibt, die nicht von einem IVD-Produkt stammen: Wird die Zweckbestimmung dann trotzdem im Wesentlichen durch IVD-Informationen bestimmt? Wie stark beeinflussen die „Nicht-IVD-Informationen“ denn die Zweckbestimmung?
Ergebnis: Sind Sie im Entscheidungspfad unten beim „Ergebnis IVDR“ angelangt?
Dann ist Ihre Software höchstwahrscheinlich ein IVD oder Teil eines IVDs, das unter die IVDR fällt. Leider hat natürlich jede Frage und jede Definition Platz für verschiedene Auslegungen. Insofern sollten Sie diese Entscheidung nicht voreilig treffen und sich im Zweifel professionelle Beratung zu diesem Thema einholen.
Hinweis: Unsere Grafik dient nur der Veranschaulichung. Für die finale Qualifizierung des Produkts empfehlen wir Ihnen, mit der englischen Version der MDCG-Guidance im Original-Wortlaut zu arbeiten. Sie können uns auch gerne kontaktieren, falls Sie Input für die Qualifizierung oder Klassifizierung Ihres Produkts benötigen. Wir entwickeln IVDR- und MDR-Software auf Auftragsbasis und geben gerne praktische Insights weiter.
1.6 Qualifizierung: Beispiele für IVDR und Nicht-IVDR
Um das Thema etwas greifbarer zu machen, finden Sie hier einige Beispiele für Software-Produkte, die laut MDCG-Guidances tendenziell …
- … ein IVD nach IVDR sind.
- … kein IVD sind, aber ein Medizinprodukt nach MDR sind.
- … weder IVD noch Medizinprodukt sind.
Qualifizierungsbeispiel: IVD nach IVDR
Ein deutliches Beispiel für ein IVD ist Software, die Rohdaten aus Labortests automatisch auswertet. Zum Beispiel:
- Software, welche die optische Dichte eines ELISA-Lesegeräts analysiert. Bei einem ELISA entsteht durch eine chemische Reaktion in der Probe ein Farbwechsel. Das Gerät schickt Licht durch die Probe und misst, wie viel Licht absorbiert wird. Aus dieser Farbintensität kann abgeleitet werden, wie viel von einem bestimmten Stoff im Blut vorhanden ist.
- Software, die Muster eines sogenannten Blots interpretieren. Ein Blot ist ein Labortest, bei dem Proteine auf eine Membran übertragen werden und dort sichtbare Linien oder Punkte bilden. Diese Muster zeigen an, ob bestimmte Antikörper oder Eiweiße vorhanden sind.
Solche Software nimmt die Messdaten auf und berechnet daraus mithilfe von medizinischen Algorithmen einen diagnostischen oder prognostischen Befund. Deshalb gilt sie in der Regel als IVD-Software.
Ein weiteres spannendes Beispiel, das wir bei QuickBird Medical gut kennen, ist die App „Accu-Chek SugarView“ von Roche. Die Accu-Chek SugarView App bestimmt den Blutzuckerbereich, indem sie den Accu-Chek Active Teststreifen und zwei Fotos, die mit der Smartphone-Kamera vom Teststreifen aufgenommen werden, auswertet. Die intelligente Software funktioniert ohne jegliches zusätzliches Glukosemessgerät. Sie wurde als IVD-Software klassifiziert und hat die CE-Kennzeichnung erhalten (mehr dazu finden Sie hier)
Qualifizierungsbeispiel: Kein IVD, sondern Medizinprodukt nach MDR
Es gibt auch Software, die zwar einen medizinischen Zweck erfüllt, aber keine Laborproben auswertet. Solche Anwendungen fallen nicht unter die IVDR, sondern unter die MDR. Dazu gehören insbesondere Programme, die:
- medizinische Bilder verarbeiten oder analysieren, zum Beispiel in der Radiologie oder Dermatologie
- Messwerte eines medizinischen Geräts bewerten, solange diese Werte nicht aus In-Vitro-Tests stammen
Wenn die Software keine Blut-, Urin- oder Gewebeproben untersucht, aber trotzdem medizinische Entscheidungen unterstützt, handelt es sich dann eben womöglich um ein Medizinprodukt nach MDR und nicht um ein IVD.
Qualifizierungsbeispiel: Kein IVD und kein MDR-Medizinprodukt
Viele Softwareprodukte erfüllen weder die Definition eines IVD nach IVDR noch die eines Medizinprodukts nach MDR. Darunter fallen zum Beispiel:
- Laborinformationssysteme (LIS), die nur Daten verwalten oder Laborprozesse organisieren
- Programme, die Ergebnisse einfach darstellen, zum Beispiel in Form von Diagrammen oder einfachen Statistiken
- Software, die Messdaten lediglich speichert oder weiterleitet, ohne diese zu interpretieren oder zu analysieren
Solche Software trifft keine eigenen diagnostischen Aussagen und hat keinen direkten medizinischen Zweck. Deshalb fällt sie weder unter die IVDR noch unter die MDR.
2. Klassifizierung: Unter welche Risikoklasse fällt Ihre Software?
„Ja, ich will!“ – Können Sie diese Aussage mit Selbstbewusstsein in Bezug auf die IVD-Qualifizierung (siehe oben) machen? Fallen Sie also in den Anwendungsbereich der IVDR? (für viele Hersteller vermutlich kein „Ich will“, sondern eher ein „Ja, ich muss 😔“). Dann wird es nun Zeit, sich mit den Risikoklassen der IVDR zu beschäftigen.
Um die regulatorischen Anforderungen für Sie als Hersteller und Ihr Produkt zu bestimmen, müssen Sie erstmal wissen, in welche Risikoklasse Ihr Produkt bzw. die Komponenten Ihres Produkts fallen.
2.1 Welche Risikoklassen gibt es unter IVDR?
Die IVDR unterscheidet zwischen den folgenden Risikoklassen: A, B, C, D
Die Idee ist, dass man Produkten mit höherem Risiko (z.B. B, C oder D) auch strengere Anforderungen auferlegt als Produkten mit sehr geringem Risiko (Klasse A).
Hinweis: Damit unterscheidet sich die Risikoklassifizierung unter IVDR von Medizinprodukten, die unter die Medical Device Regulation (MDR) fallen. Die MDR unterteilt Medizinprodukte in die Risikoklassen I, IIa, IIb und III. Mehr dazu in unserem MDR-Leitfaden.
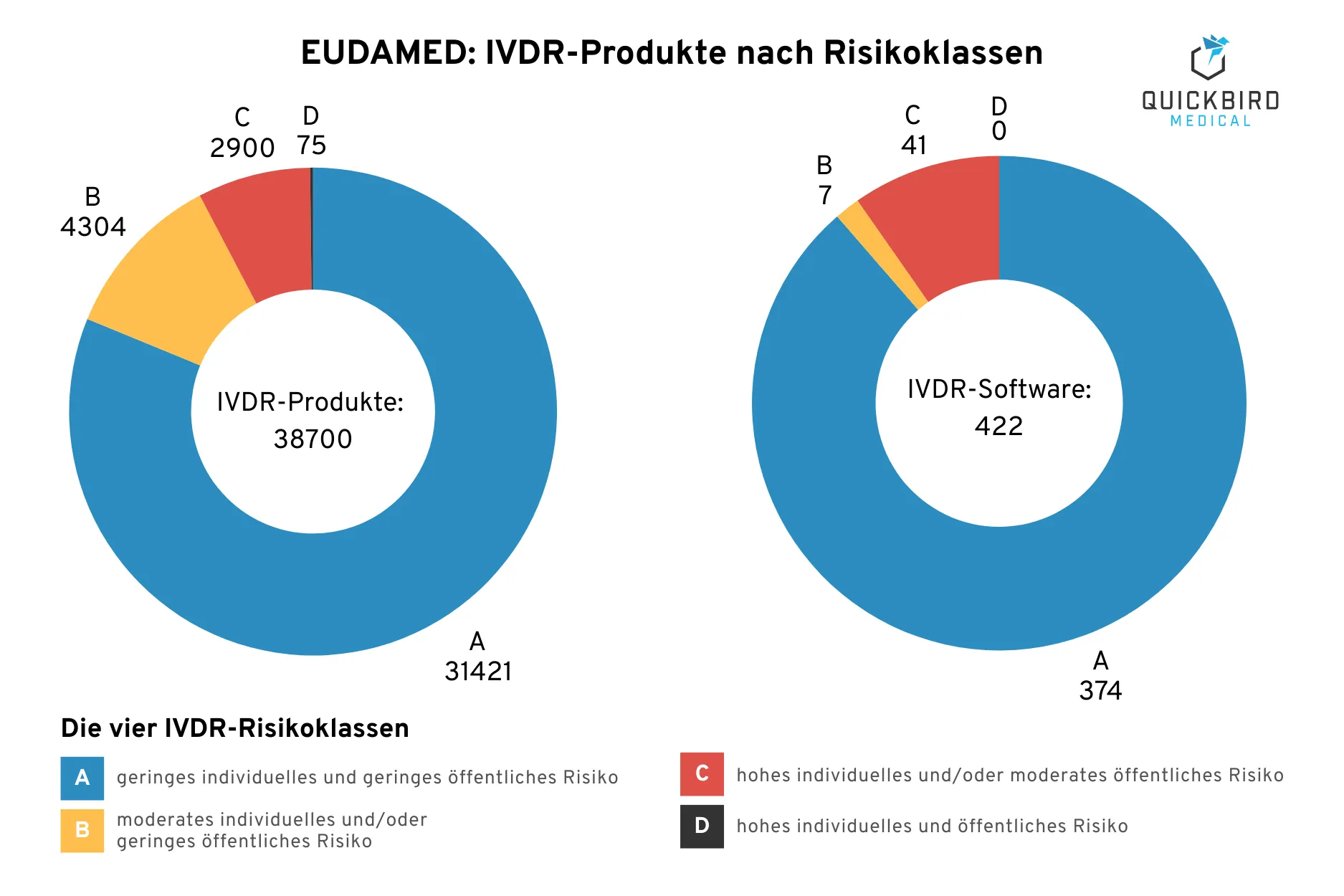
Recherche in der EUDAMED-Datenbank: Anzahl der IVDR-Produkte & IVDR-Software-Produkte pro Risikoklasse
(Beschreibung der Risiken jeweils nach IMDRF-Guidance)
In der Abbildung finden Sie das Ergebnis unserer Recherchen zur Risikoklassen-Verteilung von IVDR-Produkten innerhalb der EUDAMED-Datenbank. Vorsicht: Die Statistik hat keinerlei Aussagekraft in Bezug auf Ihr Produkt. Die Tatsache, dass die meisten Produkte in Klasse A eingeordnet sind, hat viele Gründe. Sie müssen trotzdem strukturiert evaluieren, in welche Risikoklasse Ihre Software fällt (und tatsächlich ist es gerade bei diagnostischer Software vermutlich nicht die Klasse A oder B).
2.2 Die wichtigsten Implikationen der Risikoklassen der IVDR
Die Risikoklasse eines IVD-Software-Produkts wirkt sich in vielerlei Hinsicht auf Entwicklung, Zulassungsprozess und Post-Market-Aktivitäten aus. Hier fassen wir die zentralen Aspekte für Sie zusammen.
Deckungsgleiche Anforderungen an das Qualitätsmanagement & technische Dokumentation
Je nach Risikoklasse, werden unterschiedlich strenge Anforderungen im Rahmen der IVDR gestellt. Dies betrifft aber tatsächlich nicht die sogenannten Grundlegenden Sicherheits- und Leistungsanforderungen an das IVD-Produkt. Die grundlegenden Anforderungen an das Qualitätsmanagementsystem des Herstellers und die technische Dokumentation des Produkts sind bei allen Risikoklassen (A, B, C und D) identisch (bei den höheren Risikoklassen C & D greifen aber vereinzelt Zusatzanforderungen).
Hierbei sind besonders relevant:
- Qualitätsmanagement nach ISO 13485 → Unser Leitfaden zur ISO 13485, speziell für Software-Produkte
- Software-Lebenszyklus nach IEC 62304 → Unser Leitfaden zur IEC 62304
- Risikomanagement nach ISO 14971
- Gebrauchstauglichkeit nach IEC 62366-1
Benannte Stelle ab Risikoklasse B
Der wohl größte Unterschied ist, dass Hersteller von Produkten der Klasse B, C oder D (im Gegensatz zu den meisten Produkten der Klasse A) durch einen Zertifizierungsprozess mit einer benannten Stelle gehen müssen, bevor die Produkte auf den Markt gelangen können. Eine akkreditierte Stelle muss also u.a. die technische Dokumentation und das Qualitätsmanagementsystem prüfen und freigeben.
Das ist übrigens sehr ähnlich zur Regelung unter Medical Device Regulation (MDR), wo Klasse I-Produkte quasi vom Hersteller selbst zertifiziert werden und ohne externe Prüfung auf den Markt gehen können (im Gegensatz zu Klasse IIa, IIb und III unter MDR).
Unterschiede zwischen Klasse B, C und D
Der Unterschied zwischen Risikoklasse A und B ist somit klar (die Prüfung der benannten Stelle). Gibt es ab Risikoklasse B dann jedoch keine weiteren Anforderungen mit steigender Risikoklasse (C und D)? Wofür dann 4 Klassen und nicht nur eine?
Antwort: Doch, die gibt es. Vereinfacht gesprochen, bei Risikoklasse C und D:
- Es werden vereinzelt weitere Dokumentationsanforderungen gestellt (z. B. ein zusätzlicher Kurzbericht über Sicherheit und Leistung).
- Der Detailgrad der Prüfung durch die benannte Stelle steigt mit steigender Risikoklasse.
- Bei Risikoklasse D wird ein Referenzlabor der Europäischen Union in die Prüfungen und Zulassung einbezogen.
Eine gute Übersicht bekommen Sie auch, wenn Sie im offiziellen Gesetzestext nach dem Stichwort „Klasse“ suchen und einmal alle Erwähnungen überprüfen.
2.3 Einordnung in die richtige Klasse: Regeln der IVDR
Wichtig ist zuallererst, dass die gesamte Klassifizierung auf Basis der Zweckbestimmung Ihres Produkts stattfinden muss. Wie genau Sie die Zweckbestimmung für ein Medizinprodukt definieren, erklären wir in unserem gesonderten Leitfaden hier.
Die Klassifizierung von IVDR-Produkten wird durch eine überraschend übersichtliche, kurze Liste von 7 Regeln bestimmt (ANHANG VIII der IVDR). Statt diese hier redundant in einigen Worten wiederzugeben, empfehlen wir Ihnen stark, sich einfach selbst einmal den kurzen Abschnitt in der IVDR hierzu durchzulesen (siehe unten). Wir haben den genauen Wortlaut aus der Verordnung für Sie im nächsten Kapitel eingefügt. Das ist tatsächlich auch der einzige Abschnitt der IVDR, der sich mit der Einordnung in die Risikoklasse beschäftigt.
2.3.1 Auszug der EU-Verordnung: Klassifizierungsregeln nach Anhang VIII der IVDR
Die folgenden Klassifizierungsregeln finden Sie in der Verordnung über In-Vitro-Diagnostika (IVDR) in Anhang VIII:
2. KLASSIFIZIERUNGSREGELN
2.1. Regel 1
Produkte mit den folgenden Zweckbestimmungen werden der Klasse D zugeordnet:
— Nachweis des Vorhandenseins von oder der Exposition gegenüber übertragbaren Erregern in Blut, Blutbestandteilen, Zellen, Geweben oder Organen oder in einem ihrer Derivate, um ihre Eignung für die Transfusion, Transplantation oder Zellgabe zu bewerten;
— Nachweis des Vorhandenseins von oder der Exposition gegenüber übertragbaren Erregern, die eine lebensbedrohende Krankheit mit einem hohen oder mutmaßlich hohen Verbreitungsrisiko verursachen;
— Bestimmung der Infektionslast einer lebensbedrohenden Krankheit, dessen Überwachung im Rahmen des Patientenmanagements von entscheidender Bedeutung ist.2.2. Regel 2
Produkte, die zur Blutgruppenbestimmung oder zur Feststellung einer feto-maternalen Blutgruppen-Inkompatibilität oder zur Gewebetypisierung verwendet werden, um die Immunkompatibilität von für die Transfusion, Transplantation oder Zellgabe bestimmtem Blut, Blutbestandteilen, Zellen, Geweben oder Organen festzustellen, werden der Klasse C zugeordnet, es sei denn, sie werden zur Bestimmung eines der folgenden Marker eingesetzt:
— ABNull-System [A (ABO1), B (ABO2), AB (ABO3)];
— Rhesus-System [RH1 (D), RHW1, RH2 (C), RH3 (E), RH4 (c), RH5 (e)];
— Kell-System [Kel1 (K)];
— Kidd-System [JK1 (Jka), JK2 (Jkb)];
— Duffy-System [FY1 (Fya), FY2 (Fyb)].
In diesem Fall werden sie der Klasse D zugeordnet.2.3. Regel 3
Produkte werden der Klasse C zugeordnet, wenn sie folgende Zweckbestimmung haben:
a) Nachweis des Vorhandenseins von oder Exposition gegenüber einem sexuell übertragbaren Erreger;
b) Nachweis eines Infektionserregers ohne hohes oder mutmaßlich hohes Verbreitungsrisiko im Liquor oder im Blut;
c) Nachweis eines Infektionserregers, wenn ein signifikantes Risiko besteht, dass ein fehlerhaftes Ergebnis den Tod oder eine ernste gesundheitliche Schädigung der getesteten Person, des getesteten Fötus oder Embryos oder der Nachkommen der getesteten Person verursacht;
d) Feststellung des Immunstatus von Frauen gegenüber übertragbaren Erregern zum Zwecke des pränatalen Screenings;
e) Feststellung des Vorliegens einer Infektionskrankheit oder des Immunstatus, wenn das Risiko besteht, dass ein fehlerhaftes Ergebnis zu einer Patientenmanagemententscheidung führen würde, die eine lebensbedrohende Situation für den Patienten oder die Nachkommen des Patienten schafft;
f) Einsatz als therapiebegleitende Diagnostika;
g) Einsatz zur Stadieneinteilung einer Krankheit, wenn das Risiko besteht, dass ein fehlerhaftes Ergebnis zu einer Patientenmanagemententscheidung führen würde, die eine lebensbedrohende Situation für den Patienten oder die Nachkommen des Patienten schafft;
h) Einsatz zur Krebsvorsorge, -diagnose oder -stadieneinteilung;
i) Durchführung von Gentests beim Menschen;
j) Überwachung des Niveaus von Arzneimitteln, Stoffen oder biologischen Komponenten, wenn das Risiko besteht, dass ein fehlerhaftes Ergebnis zu einer Patientenmanagemententscheidung führen würde, die eine lebensbedrohende Situation für den Patienten oder die Nachkommen des Patienten schafft;
k) Management von Patienten, die an einer lebensbedrohenden Krankheit leiden oder deren Zustand lebensbedrohend ist;
l) Untersuchung auf genetisch bedingte Störungen beim Embryo oder Fötus;
m) Untersuchung auf genetisch bedingte Störungen bei Neugeborenen, wenn der fehlende Nachweis und die fehlende Behandlung dieser Störungen zu lebensbedrohenden Situationen oder ernsten gesundheitlichen Schädigungen führen können.2.4. Regel 4
a) Produkte zur Eigenanwendung werden der Klasse C zugeordnet, ausgenommen Produkte zur Feststellung einer Schwangerschaft, zur Fertilitätsuntersuchung und zur Bestimmung des Cholesterinspiegels und Produkte zum Nachweis von Glukose, Erythrozyten, Leukozyten und Bakterien im Urin, die der Klasse B zugeordnet werden.
b) Produkte zur Verwendung in patientennahen Tests werden für sich allein klassifiziert.2.5. Regel 5
Die folgenden Produkte werden der Klasse A zugeordnet:
a) Erzeugnisse für den allgemeinen Laborbedarf, Zubehör ohne kritische Merkmale, Pufferlösungen, Waschlösungen sowie allgemeine Nährmedien und histologische Färbungen, die vom Hersteller dafür vorgesehen sind, die Produkte für In-vitro-Diagnoseverfahren im Zusammenhang mit einer spezifischen Untersuchung einsetzbar zu machen;
b) Instrumente, die vom Hersteller speziell für die Verwendung bei In-vitro-Diagnoseverfahren vorgesehen sind;
c) Probenbehältnisse.2.6. Regel 6
Produkte, die nicht unter die zuvor beschriebenen Klassifizierungsregeln fallen, werden der Klasse B zugeordnet.2.7. Regel 7
Produkte, bei denen es sich um Kontrollen ohne einen zugewiesenen quantitativen oder qualitativen Wert handelt, werden der Klasse B zugeordnet.
2.3.2 Auszug der EU-Verordnung: Durchführungsvorschriften für die Klassifizierung
Neben den Klassifizierungsregeln finden Sie in der IVDR noch einige Durchführungsvorschriften, welche die Klassifizierung betreffen. Untenstehend finden Sie den Auszug aus der IVDR im Anhang VIII.
1. DURCHFÜHRUNGSVORSCHRIFTEN
1.1. Die Anwendung der Klassifizierungsregeln richtet sich nach der Zweckbestimmung der Produkte.
1.2. Wenn das betreffende Produkt dazu bestimmt ist, in Verbindung mit einem anderen Produkt angewandt zu werden, werden die Klassifizierungsregeln auf jedes Produkt gesondert angewendet.
1.3. Zubehör für ein In-vitro-Diagnostikum wird unabhängig von dem Produkt, mit dem es verwendet wird, gesondert klassifiziert.
1.4. Software, die ein Produkt steuert oder dessen Anwendung beeinflusst, wird derselben Klasse zugerechnet wie das Produkt. Ist die Software von anderen Produkten unabhängig, so wird sie für sich allein klassifiziert.
1.5. Kalibratoren zur Verwendung mit einem Produkt werden derselben Klasse zugerechnet wie das Produkt.
1.6. Kontrollmaterialien mit quantitativen oder qualitativen zugeordneten Werten, die für einen spezifischen oder mehrere Analyten bestimmt sind, werden derselben Klasse zugerechnet wie das Produkt.
1.7. Der Hersteller berücksichtigt alle Klassifizierungs- und Durchführungsregeln, um die angemessene Einstufung des Produkts festzustellen.
1.8. Wenn der Hersteller für ein Produkt mehrere Zweckbestimmungen angibt und das Produkt daher mehr als einer Klasse zuzuordnen ist, wird es in die höchste der Klassen eingestuft.
1.9. Für den Fall, dass für dasselbe Produkt mehrere Klassifizierungsregeln gelten, wird die Regel der Einstufung in die höchste dieser Klassen angewendet.
1.10. Jede Klassifizierungsregel gilt für erstmalige Tests, Bestätigungstests und Ergänzungstests.
2.4 MDCG-Guidance zur Klassifizierung nach IVDR
Nicht jeder Satz der oben gezeigten Klassifizierungsregeln der IVDR ist selbsterklärend und eindeutig. Daher gibt es zusätzliche Leitfäden der MDCG, die Ihnen u.a. bei der Bestimmung der Risikoklasse helfen sollen.
Für die Klassifizierung von Software-Produkten gibt es zwei relevante MDCG-Guidance-Dokumente:
- Allgemeine Guidance – „MDCG 2020-16: Guidance on Classification Rules for in vitro Diagnostic Medical Devices under
Regulation (EU) 2017/746“: Dieses Dokument liefert zu jeder Klassifizierungsregel der IVDR hilfreiche Beispiele und Anmerkungen. So erhalten Sie zusätzliche Unterstützung bei der Evaluation, ob bestimmte Klassifizierungsregeln auf Ihr Produkt anwendbar sind. –> Link - Software-spezifisch für MDR und IVDR – „MDCG 2019-11: Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 IVDR“: Das Dokument geht u.a. auf die Besonderheiten der Qualifizierung und Klassifizierung von Software ein. Im Gegensatz zur MDCG 2020-16 ist der Umfang der relevanten Abschnitte verhältnismäßig knapp. –> Link
Hinweis: Die Formulierung „Regulation (EU) 2017/746“ steht für die IVDR. „Regulation (EU) 2017/745“ steht hingegen für die MDR.
2.5 Klassifizierung: Software-Beispiele für alle Klassen der IVDR
Auch für die Klassifizierung möchten wir Ihnen einige Software-Beispiele aus der MDCG-Guidance an die Hand geben, um die Thematik zu verdeutlichen. Natürlich sind dies nur Veranschaulichungen und der Teufel steckt manchmal im Detail. Insofern sollten Sie die Klassifizierung Ihres Produkts nicht rein auf dem Vergleich mit existierenden Beispielen vornehmen, sondern alle Regeln der IVDR einzeln prüfen.
Software-Beispiel für Risikoklasse A
Eine Software gehört vor allem zur Risikoklasse A, wenn sie ausschließlich dazu gedacht ist, ein IVD-Gerät (nach IVDR) zu steuern, welches ebenfalls in Klasse A eingestuft ist. Das ergibt sich aus Anhang VIII 1.4 der IVDR: „Software, die ein Produkt steuert oder dessen Anwendung beeinflusst, wird derselben Klasse zugerechnet wie das Produkt.“
Ein konkretes Beispiel ist eine Software, die einen CRP-Analyser (C-reaktives-Protein-Gerät), der unter die Klasse A fällt, aus der Ferne bedient. Die Software trifft keine eigenen diagnostischen Aussagen, sondern sorgt nur dafür, dass das Gerät funktioniert. Deshalb wird sie in dieselbe Klasse wie das Gerät eingestuft, hier also Klasse A.
Software-Beispiel für Risikoklasse B
Etwas schwierig ist es, offizielle Beispiele für Software der Klasse B zu finden. (Software-)Produkte landen nur in Klasse B, falls sie in spezielle Produktkategorien zur Eigenanwendung (z.B. Schwangerschaftstests) fallen (Regel 4) oder sonst keine der anderen Klassifizierungsregeln greift (Regel 6).
In der Guidance „MDCG 2020-16¡ findet sich folgendes Beispiel:
„Blood gas analyser and the software driving and influencing it are classified as […] Class B (Rule 6) where the blood gas analyser directly measures a parameter from a specimen (in the absence of a reagent or disposable sensor cassette) AND where an erroneous result in a measured parameter WILL NOT LEAD TO a patient management decision resulting in a lifethreatening situation.“
Software-Beispiel für Risikoklasse C
Software, die in einem automatisierten ELISA-System HbA1c-Werte berechnet und zur Diabetes-Diagnose oder Verlaufskontrolle verwendet wird, fällt unter die Klasse C. Das lässt sich aus Regel 3(k) der IVDR ableiten.
Software, die in einem automatisierten PAP-Screening-System Abstriche als auffällig oder unauffällig klassifiziert, fällt auch unter Klasse C. Das lässt sich aus Regel 3(h) der IVDR ableiten.
Software-Beispiel für Risikoklasse D
Software, die die Ergebnisse eines automatisierten Line-Immunoassays für HIV-Antikörper interpretiert, fällt unter die Klasse D. Das lässt sich aus Regel 1 der IVDR ableiten. In diesem Fall bewertet die Software hochkritische Infektionsmarker.
3. Wer trifft die finale Entscheidung zur Qualifizierung & Klassifizierung?
Sie als Hersteller des Produkts müssen diese Entscheidung treffen. Die Verantwortung dafür, die Anwendbarkeit der IVDR auf Ihr (Software-)Produkt zu prüfen (Qualifizierung) sowie die Einstufung in die passende Risikoklasse, liegt initial komplett bei Ihnen.
Dabei ist eine sorgfältige Bewertung entscheidend, denn eine benannte Stelle kann die Zertifizierung ablehnen, wenn diese zu einer anderen Einschätzung bei der Risikoklassifizierung kommt. Außerdem sind Sie natürlich rechtlich angreifbar, falls Sie diese Einstufung nicht richtig vornehmen.
Gerade bei einer Einstufung in die Risikoklasse A sollten Sie vorsichtig sein, da die Klassifizierung hier initial nicht von einer benannten Stelle geprüft wird. Somit haben Sie noch weniger gefühlte Sicherheit bei der Auswahl der Risikoklasse.
Außerdem: Obwohl hier keine benannte Stelle eingebunden wird, kann die zuständige Aufsichtsbehörde eine fehlerhafte Klassifizierung bemerken und im schlimmsten Fall verlangen, dass das Produkt vom Markt genommen wird. Auch Produkte der Risikoklasse A werden in unregelmäßigen Abständen Prüfungen unterzogen. Des Weiteren wird im Falle eines Personenschadens durch Ihr Produkt sicherlich auch rechtlich hinterfragt, ob die von Ihnen als Hersteller zugewiesene Risikoklasse angemessen war.
Wenn Sie unsicher sind, lohnt es sich, eine unabhängige Einschätzung einzuholen. Mögliche Anlaufstellen sind:
- ein spezialisierter Rechtsanwalt, der ein Gutachten erstellen kann
- ein Regulatory-Expert oder Berater mit entsprechender Erfahrung
- das BfArM (in der Praxis aufgrund monatelanger Bearbeitungszeiten allerdings schwierig)
QuickBird Medical entwickelt IVDR- und MDR-Software für Kunden auf Auftragsbasis. Gerne geben wir Ihnen auf Basis unserer Erfahrung Input zur Klassifizierungs- und Qualifizierungsentscheidung. Bei Bedarf können wir Ihnen z. B. auch einen Anwalt aus unserem Kontaktkreis vermitteln, der für Sie ein Gutachten erstellen kann.
→ Kontakt aufnehmen
Wichtig: Weder eine externe Einschätzung noch ein formelles Gutachten bieten eine vollständige rechtliche Absicherung. Sie können im Falle eines Rechtsstreits jedoch als wichtige Entscheidungsgrundlage dienen.
4. Gemeinsamkeiten und Unterschiede: MDR vs. IVDR
Die MDR regelt klassische Medizinprodukte, die meist direkt am Patienten eingesetzt werden, während die IVDR In-vitro-Diagnostika betrifft, also Produkte, die z. B. Proben wie Blut oder Gewebe untersuchen.
Beide Verordnungen funktionieren in vielen Bereichen ähnlich: Hersteller brauchen ein Qualitätsmanagementsystem, eine technische Dokumentation, eine verantwortliche Person für die Regelkonformität und müssen ihre Produkte nach festen Risikoklassen einstufen.
Größere Unterschiede liegen u.a. in den folgenden Punkten:
- Unterschiedliche Klassifizierungslogik: Die Risikoklassen und die Klassifizierungslogik der IVDR und MDR unterscheiden sich stark. Hier ist auch zu beachten, dass Software-Produkte unter IVDR deutlich seltener in die niedrigste Klasse (A) fallen, als dies bei MDR mit Klasse I der Fall ist. Somit ist bei den meisten IVD-Produkten mit Software eine benannte Stelle involviert.
- Spezifischere Leistungsbewertung bei IVDs: IVD-Produkte müssen eine sehr umfangreiche Performance Evaluation durchlaufen. Diese umfasst unter anderem die wissenschaftliche Validität, analytische Performance (z. B. Sensitivität, Spezifität) und die klinische Performance. Auch unter MDR gibt es Medizinprodukte, die derartige Validierungsanforderungen einhalten müssen (z. B. ein Produkt zur Diagnose von Krankheiten auf Basis von CT-Bilddaten). Diese Anforderungen sind in der IVDR jedoch deutlich konkreter definiert als in der MDR.
- Zusätzliche Prüfmechanismen bei Hochrisiko-IVDs: Produkte der IVDR-Klasse D (z. B. HIV- oder Hepatitis-Tests) müssen durch EU-Referenzlabore (EURLs) unabhängig überprüft werden.
5. Fazit
IVDR-Software bewegt sich im Spannungsfeld zwischen technischer Entwicklung, Laborrealität und strengen regulatorischen Anforderungen.
Dieser Artikel soll Ihnen helfen, die ersten zentralen Fragen zu beantworten: Fällt Ihre Software überhaupt unter die IVDR und in welche Risikoklasse gehört sie? Wenn diese Eckpfeiler stehen, haben Sie eine solide Basis, um Qualitätsmanagement, die weitere Dokumentation, Validierung und Zusammenarbeit mit einer benannten Stelle gezielt anzugehen.
Im Zuge der IVDR ist zu beachten, dass der Großteil aller Software-basierten Diagnose-Produkte nicht in Risikoklasse A fallen wird. Daher sollten Sie damit rechnen, dass Sie als Hersteller eine benannte in den Zulassungsprozess inkludieren müssen. Die Kosten und den zusätzlichen Zeitaufwand für diese Zertifizierung sollten Sie in Ihrer Produktplanung berücksichtigen.
Über uns: QuickBird Medical entwickelt IVDR- und MDR-Software für Kunden auf Auftragsbasis. Wir übernehmen sowohl die technische Umsetzung als auch die regulatorische Konformität Ihres (Software-)Produkts. Wenn Sie also ein IVDR- oder MDR-Produkt planen, welches Software-Komponenten enthält, kontaktieren Sie uns gern für ein unverbindliches Erstgespräch.
→ Kontakt aufnehmen
In Zukunft planen wir weitere Artikel, die Ihnen bei der Planung und Entwicklung von IVDR-Produkten bzw. speziell der Software-Komponente dieser helfen sollen. In unserem Newsletter informieren wir Sie über neue Artikel und Änderungen zu diesem Thema.



