

Seit 2019 haben rund 73 Millionen gesetzlich Versicherte in Deutschland Anspruch auf digitale Gesundheitsanwendungen (DiGA): die sogenannte „App auf Rezept“. Doch bei angehenden DiGA-Herstellern sorgt das Zusammenspiel von DiGA und Medizinprodukte-Regulierung häufig für Verwirrung. Die Anforderungen von Medizinprodukten und DiGA sind sehr unterschiedlich, haben gleichzeitig aber viele Überschneidungen. Diese Schnittstelle gilt es genau zu verstehen.
Dieser Artikel klärt wichtige Fragen zum Verhältnis zwischen DiGA und Medizinprodukten:
- Muss eine DiGA ein Medizinprodukt sein?
- Welche Risikoklassen sind für DiGA erlaubt?
- Was ist der Zusammenhang zwischen DiGA-Status und MDR?
- Welche Zusatzanforderungen müssen DiGA erfüllen?
- Darf eine DiGA mit Hardware-Medizinprodukten kombiniert werden?
Der Artikel richtet sich an angehende DiGA-Hersteller und andere Fachkreise, die verstehen wollen, welche regulatorischen Anforderungen für digitale Gesundheitsanwendungen gelten. Wir bei QuickBird Medical waren bereits in die Entwicklung von über 15 DiGA auf Auftragsbasis involviert und geben in Fachartikeln unser Wissen weiter.
Inhaltsverzeichnis
- 1. Zusammenspiel: DiGA und Medizinprodukt
- 2. Welche MDR-Medizinprodukt-Klassen sind für DiGA erlaubt?
- 3. Medizinprodukt vs. DiGA: Zusatzanforderungen für DiGA
- 4. Anbindung von medizinischen Geräten & Hardware an DiGA
- 5. Unterschiede zwischen Medizinprodukt und DiGA
- 6. Fazit: Empfehlungen für Hersteller
1. Zusammenspiel: DiGA und Medizinprodukt
Mit dem Konzept der DiGA wurde ein strukturierter Pfad geschaffen, um patientenorientierte medizinische Software und Apps in die Erstattung der gesetzlichen Krankenversicherung in Deutschland zu bringen.
Voraussetzung dafür: Die Anwendung muss zunächst als Medizinprodukt nach der europäischen Medical Device Regulation (MDR) auf den Markt gebracht werden. Als Medizinprodukt gilt Software, die vom Hersteller für einen medizinischen Zweck bestimmt ist: etwa zur Diagnose, Therapie oder Überwachung von Krankheiten. Erst mit gültiger CE-Kennzeichnung kann ein Hersteller den DiGA-Antrag beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) stellen.
Zusätzlich zur Zulassung als Medizinprodukt, muss eine medizinische Anwendung aber auch eine breite Palette weiterer Anforderungen erfüllen, um in das DiGA-Verzeichnis aufgenommen zu werden.
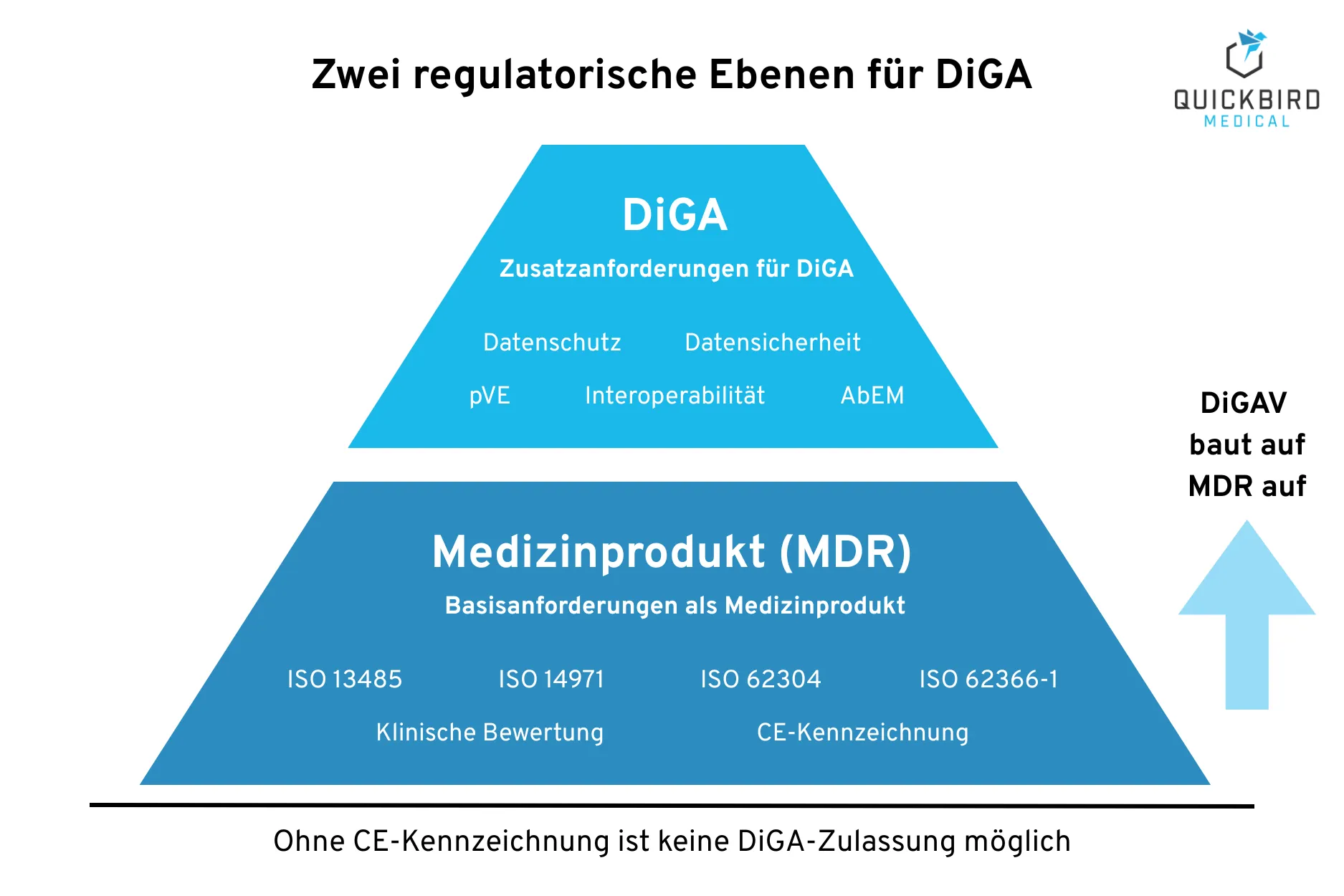
Anforderungen von DiGA und Software-Medizinprodukten
1.1 Muss eine DiGA also ein Medizinprodukt sein?
Die Antwort ist eindeutig: Ja, jede DiGA muss ein Medizinprodukt sein.
Das ergibt sich direkt aus der gesetzlichen Definition. § 33a SGB V definiert DiGA als „Medizinprodukte niedriger Risikoklasse, deren Hauptfunktion wesentlich auf digitalen Technologien beruht“. Eine DiGA ist also keine Alternative zum Medizinprodukt, sondern eine spezielle Kategorie von Medizinprodukten mit zusätzlichen Anforderungen.
1.2 Was bedeutet das konkret für Hersteller?
Bevor ein Hersteller überhaupt einen Antrag auf Aufnahme in das DiGA-Verzeichnis beim BfArM stellen kann, muss das Konformitätsbewertungsverfahren nach MDR vollständig abgeschlossen sein. Die CE-Kennzeichnung ist dabei der Nachweis, dass das Produkt die grundlegenden Sicherheits- und Leistungsanforderungen erfüllt und in Europa in Verkehr gebracht werden darf.
Wichtig zu verstehen: Das BfArM prüft im DiGA-Verfahren nicht die MDR-Konformität selbst. Diese wird vorausgesetzt und durch die CE-Kennzeichnung belegt. Das BfArM prüft stattdessen die zusätzlichen Anforderungen der DiGAV, etwa den Nachweis positiver Versorgungseffekte, Datenschutz und Interoperabilität.
1.3 Wann ist meine Software ein Medizinprodukt?
Bevor also die Frage nach dem DiGA-Status relevant wird, müssen Hersteller zunächst klären, ob ihre Software überhaupt ein Medizinprodukt ist. Diese Entscheidung ist grundlegend für den gesamten regulatorischen Weg.
Ob eine Software als Medizinprodukt gilt, hängt primär von der medizinischen Zweckbestimmung ab, die der Hersteller festlegt. Software ist nur dann ein Medizinprodukt, wenn sie einen medizinischen Zweck verfolgt, also wenn sie wie bereits erwähnt entweder die Diagnose, Therapie oder Überwachung von Krankheiten unterstützt.
Die Abgrenzung ist nicht immer trivial. Eine App, die einfach nur Fitnessdaten trackt, ist in der Regel kein Medizinprodukt. Dieselbe App mit einer Funktion zur Erkennung von Herzrhythmusstörungen hingegen schon. Entscheidend ist, welchen Zweck der Hersteller für das Produkt deklariert und welche Funktionen tatsächlich angeboten werden.
1.4 Wann ist Software kein Medizinprodukt?
Nicht jede Gesundheits-App ist automatisch ein Medizinprodukt. Folgende Kategorien fallen z. B. in der Regel nicht unter die MDR:
- Reine Datenspeicherung oder -übertragung: Software, die Gesundheitsdaten nur speichert oder weiterleitet, ohne sie medizinisch zu verarbeiten oder zu interpretieren
- Administrative Funktionen: Terminplanung, Erinnerungsfunktionen ohne medizinischen Kontext, Praxisverwaltung
- Allgemeine Gesundheitsinformationen: Apps, die generische Informationen bereitstellen, ohne individuelle Empfehlungen zu geben
- Lifestyle- und Wellness-Anwendungen: Fitness-Tracker, Ernährungs-Apps oder Meditations-Apps ohne medizinischen Zweck
Unser ausführlicher Leitfaden „Ist Ihre Software ein Medizinprodukt?“ behandelt dieses Thema im Detail mit konkreten Entscheidungshilfen.
2. Welche MDR-Medizinprodukt-Klassen sind für DiGA erlaubt?
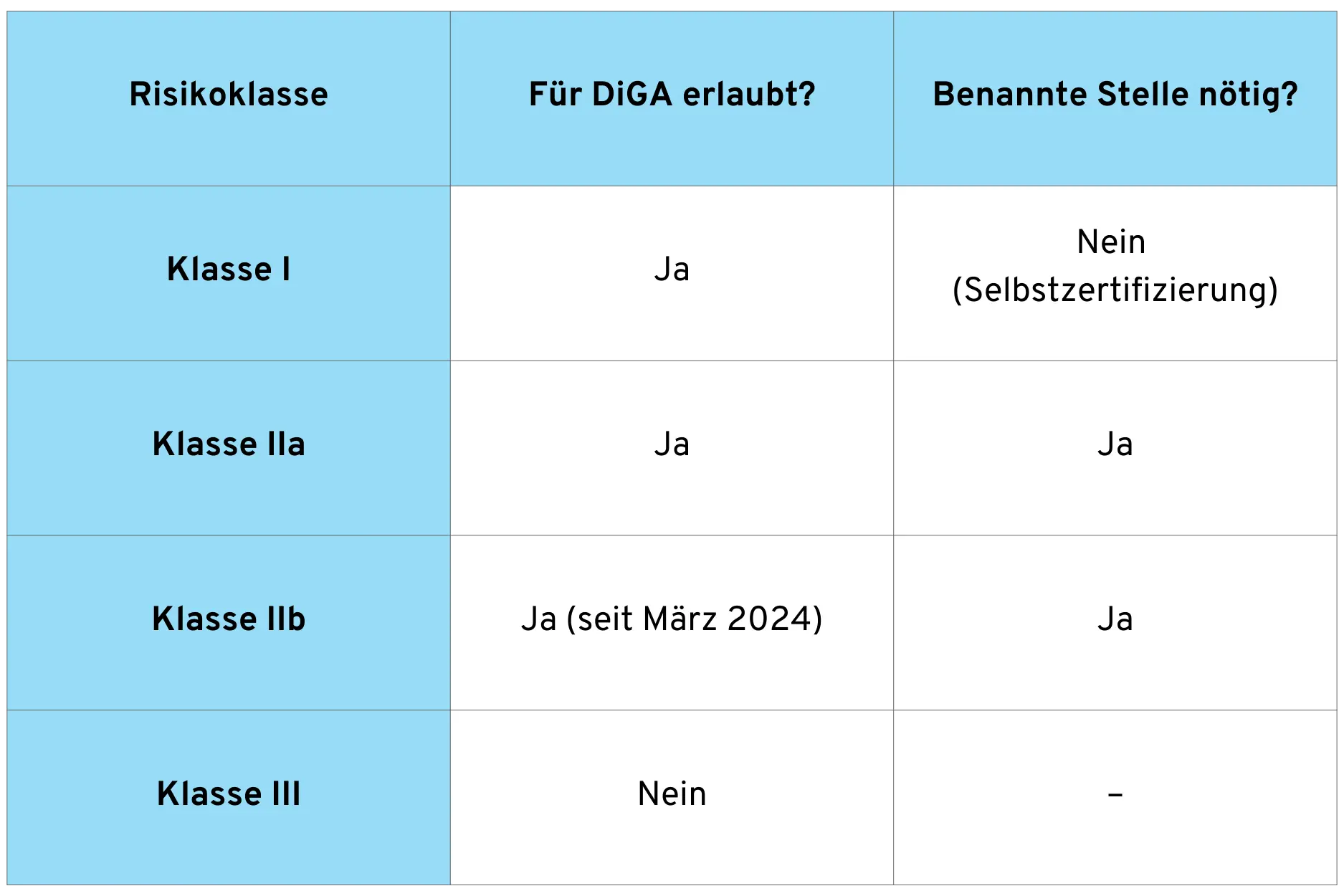
Nicht jedes Medizinprodukt kann eine DiGA werden. Der Gesetzgeber hat die DiGA-Fähigkeit auf bestimmte Risikoklassen beschränkt. Dies spiegelt die Idee wider, dass DiGA als niedrigschwelliges Versorgungsangebot mit entsprechend begrenztem Risikopotenzial konzipiert sind.
Die folgende Übersicht zeigt, welche Risikoklassen für DiGA zugelassen sind:
 (Klicken zum Vergrößern)
(Klicken zum Vergrößern)
Bei Medizinprodukten der Klasse I kann der Hersteller die Konformitätsbewertung selbst durchführen und die Konformitätserklärung ausstellen. Bei Klasse IIa und IIb ist hingegen die Einbeziehung einer Benannten Stelle erforderlich, die das Qualitätsmanagementsystem und die technische Dokumentation prüft.
2.1 Erweiterung durch das Digital-Gesetz (DigiG)
Eine wichtige Änderung trat mit dem Digital-Gesetz (DigiG) am 26. März 2024 in Kraft: Seitdem können auch Medizinprodukte der Risikoklasse IIb in das DiGA-Verzeichnis aufgenommen werden. Zuvor waren nur die Klassen I und IIa zugelassen.
Diese Erweiterung ermöglicht DiGA mit höherem Risikoprofil. Allerdings gelten für Klasse-IIb-DiGA besondere Anforderungen: Eine vorläufige Aufnahme zur Erprobung ist zum Beispiel nicht möglich. Hersteller müssen bereits bei Antragstellung die Ergebnisse einer Studie zum Nachweis eines medizinischen Nutzens vorlegen.
2.2 Wie bestimme ich die Medizinprodukt-Risikoklasse für meine DiGA?
Die korrekte Klassifizierung ist einer der ersten und wichtigsten Schritte bei der Entwicklung einer DiGA. Sie bestimmt nicht nur, ob eine Benannte Stelle einbezogen werden muss, sondern beeinflusst den gesamten regulatorischen Aufwand und die Kosten.
Die Klassifizierung von Software-Medizinprodukten erfolgt nach den Regeln in Anhang VIII der MDR. Für Software ist dabei Regel 11 besonders relevant. Diese Regel klassifiziert Software u. a. anhand des Risikos, das von den bereitgestellten Informationen für den Patienten ausgeht.
Die Klassifizierung hängt von mehreren Faktoren ab: der Art der Erkrankung (schwerwiegend vs. nicht-schwerwiegend), der Art der Entscheidungsunterstützung (Diagnose, Therapie, Überwachung) und der Reversibilität möglicher Schäden durch falsche Informationen.
Unser Artikel Klassifizierung von Software-Medizinprodukten erläutert die Klassifizierungsregeln im Detail mit praktischen Beispielen.
Praxistipp: Befassen Sie sich frühzeitig mit der Klassifizierung – idealerweise bereits in der Konzeptionsphase. Die Risikoklasse bestimmt den regulatorischen Aufwand, die Kosten und den Zeitrahmen bis zur Markteinführung maßgeblich.
3. Medizinprodukt vs. DiGA: Zusatzanforderungen für DiGA
Eine DiGA muss zwei regulatorische Ebenen erfüllen: zunächst die MDR-Anforderungen als Medizinprodukt und darüber hinaus die spezifischen Anforderungen der DiGA-Verordnung (DIGAV). Dieser Abschnitt gibt einen Überblick über beide Ebenen und zeigt, welche zusätzlichen Hürden DiGA-Hersteller im Vergleich zu „normalen“ Medizinprodukte-Herstellern nehmen müssen.
3.1 Anforderungen als Medizinprodukt (MDR)
Jede DiGA muss zunächst die grundlegenden MDR-Anforderungen erfüllen. Diese bilden das Fundament, auf dem die DiGA-spezifischen Anforderungen aufbauen. Die MDR verlangt ein umfassendes Qualitäts- und Dokumentationssystem.
Zu den zentralen Anforderungen gehören:
- Qualitätsmanagementsystem nach ISO 13485, das alle qualitätsrelevanten Prozesse dokumentiert und steuert
- Risikomanagement nach ISO 14971, das potenzielle Gefährdungen identifiziert und Maßnahmen zur Risikominimierung definiert
- Software-Lebenszyklus nach IEC 62304, der die Entwicklung und den Betrieb von Software strukturiert
- Gebrauchstauglichkeit nach IEC 62366-1, die sicherstellt, dass das Produkt sicher und effektiv bedient werden kann
- Klinische Bewertung, die den klinischen Nutzen und die Sicherheit des Produkts belegt
- Post-Market-Surveillance, die das Produkt nach dem Inverkehrbringen kontinuierlich überwacht
Diese Anforderungen gelten für alle Medizinprodukte. Dies ist unabhängig davon, ob sie zusätzlich als DiGA in die Erstattung gebracht werden sollen.
Unser Artikel Zulassung & Zertifizierung von Software-Medizinprodukten behandelt die MDR-Anforderungen ausführlich.
3.2 Zusatzanforderungen für DiGA (DiGAV)
Über die MDR-Medizinprodukt-Anforderungen hinaus definiert die DiGAV spezifische Kriterien, die eine DiGA erfüllen muss. Diese Anforderungen werden vom BfArM im Rahmen des Fast-Track-Verfahrens geprüft.
3.2.1 Positive Versorgungseffekte (pVE)
DiGA müssen einen positiven Versorgungseffekt nachweisen. Dieser kann entweder ein medizinischer Nutzen sein (z. B. Verbesserung des Gesundheitszustands) oder eine patientenrelevante Struktur- und Verfahrensverbesserung (z. B. bessere Therapieadhärenz, vereinfachter Zugang zur Versorgung).
Für die dauerhafte Aufnahme ins DiGA-Verzeichnis ist eine vergleichende Studie erforderlich, die den positiven Versorgungseffekt belegt. Bei vorläufiger Aufnahme erhält der Hersteller einen Erprobungszeitraum von bis zu 12 Monaten (in begründeten Ausnahmen bis zu 24 Monate), um diesen Nachweis zu erbringen. Auch für eine Aufnahme auf Erprobung muss aber ein umfangreiches Evaluationskonzept inklusive Pilotstudie vorgelegt werden.
Mehr dazu: Positive Versorgungseffekte nachweisen
3.2.2 Datenschutz
Die DiGAV stellt über die DSGVO hinaus besonders strenge Anforderungen an den Datenschutz. Es sind nur bestimmte, abschließend definierte Zwecke der Datenverarbeitung zulässig. Für die Verarbeitung von Gesundheitsdaten außerhalb Deutschlands gelten besondere Regelungen. Transparenz über Art und Umfang der Datenverarbeitung ist Pflicht.
Mehr dazu: DiGA Datenschutz-Anforderungen
3.2.3 Datensicherheit
Ein Informationssicherheitsmanagementsystem (ISMS) ist für DiGA-Hersteller verpflichtend und muss sogar nach ISO 27001 zertifiziert werden.
Zusätzlich müssen DiGA-Hersteller eine Zertifizierung nach BSI TR-03161 erlangen. Dies ist eine sehr anspruchsvolle Zertifizierung für die Informationssicherheit, die durch Prüfstellen und abschließend das BSI (Bundesamt für Sicherheit in der Informationstechnik) vergeben wird.
Mehr dazu: BSI TR-03161 für DiGA
3.2.4 Interoperabilität
DiGA müssen interoperable Standards nutzen, damit Gesundheitsdaten zwischen verschiedenen Systemen ausgetauscht werden können. Konkret bedeutet das: Die Möglichkeit, DiGA-Daten im menschenlesbaren und maschinenlesbaren Format exportieren zu können (durch Nutzung von FHIR-Profilen und dem MIO DiGA Toolkit) sowie die Anbindung an die elektronische Patientenakte (ePA).
Mehr dazu: Interoperabilität für DiGA
3.2.5 Qualitätsanforderungen
Die DiGAV definiert weitere Qualitätsanforderungen in verschiedenen Bereichen:
- Robustheit: Die DiGA muss stabil funktionieren und fehlertolerant sein
- Verbraucherschutz: Transparente Information, keine irreführende Werbung, Einhaltung des Heilmittelwerbegesetzes
- Nutzerfreundlichkeit: Barrierefreie Gestaltung, intuitive Bedienung
- Unterstützung der Leistungserbringenden: Sinnvolle Integration in bestehende Versorgungsprozesse
- Qualität medizinischer Inhalte: Evidenzbasierung, Aktualität, fachliche Korrektheit
- Patientensicherheit: Risikominimierende Maßnahmen, angemessene Warnhinweise
3.2.6 Anwendungsbegleitende Erfolgsmessung (AbEM)
Nach der Aufnahme in das DiGA-Verzeichnis müssen Sie kontinuierlich Daten zur Nutzung und Wirksamkeit ihrer DiGA erheben. Die anwendungsbegleitende Erfolgsmessung umfasst Nutzungsdaten, Patientenzufriedenheit (PGI-C-Skala) und perspektivisch indikationsspezifische PROMs. Diese Daten müssen regelmäßig an das BfArM berichtet werden.
3.3 Weiterführende Informationen
Mehr Informationen, welche Kriterien Sie einhalten müssen, um eine DiGA zu sein, finden Sie auch hier: DiGA Definition & Kriterien
4. Anbindung von medizinischen Geräten & Hardware an DiGA
Eine häufige Frage von Herstellern betrifft die Kombination von DiGA mit Hardware: Kann eine DiGA auch Sensoren, Wearables oder andere Medizingeräte einbinden? Die Antwort ist differenziert. Grundsätzlich ja, aber unter bestimmten Bedingungen.
DiGA sind primär Software-Medizinprodukte. Ihre Hauptfunktion muss wesentlich auf digitalen Technologien beruhen. Sie können jedoch mit Hardware-Medizinprodukten und anderen Systemen kommunizieren, Daten austauschen und diese verarbeiten – solange die digitale Hauptfunktion überwiegt.
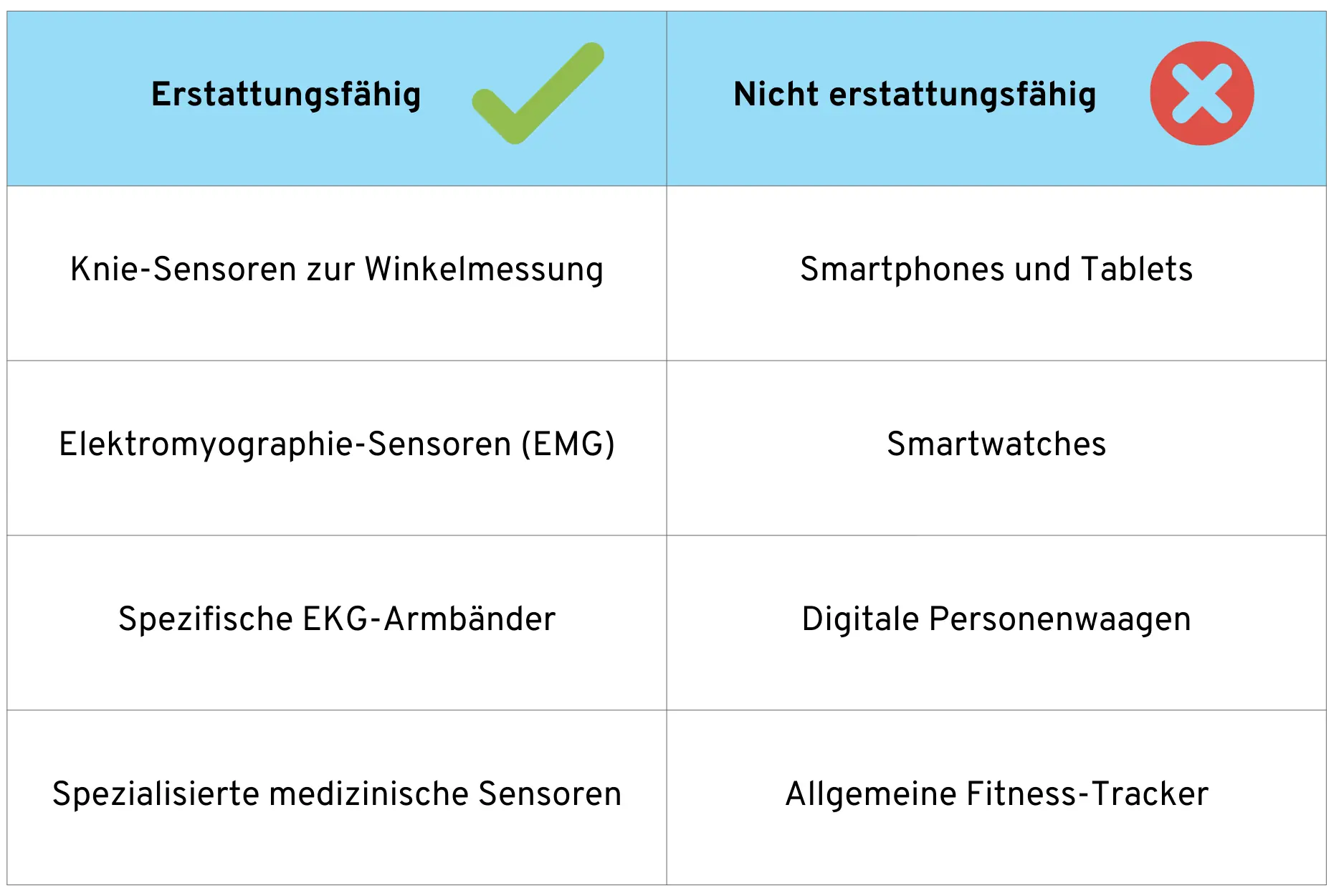
4.1 Erstattungsfähige vs. nicht erstattungsfähige Hardware
Ein wichtiger Aspekt für Hersteller ist die Frage der Erstattungsfähigkeit. Nicht jede Hardware, die mit einer DiGA kombiniert wird, wird auch von der GKV erstattet.
Die folgende Übersicht zeigt exemplarische Beispiele, welche Hardware-Typen potenziell erstattet werden und welche nicht:
 (Klicken zum Vergrößern)
(Klicken zum Vergrößern)
Grundregel: Erstattungsfähig sind spezialisierte medizinische Geräte, die keine Gegenstände des täglichen Lebens darstellen. Gebrauchsgegenstände des täglichen Lebens, auch wenn sie medizinische Funktionen haben, werden nicht von der GKV bezahlt, können aber mit einer DiGA verwendet werden.
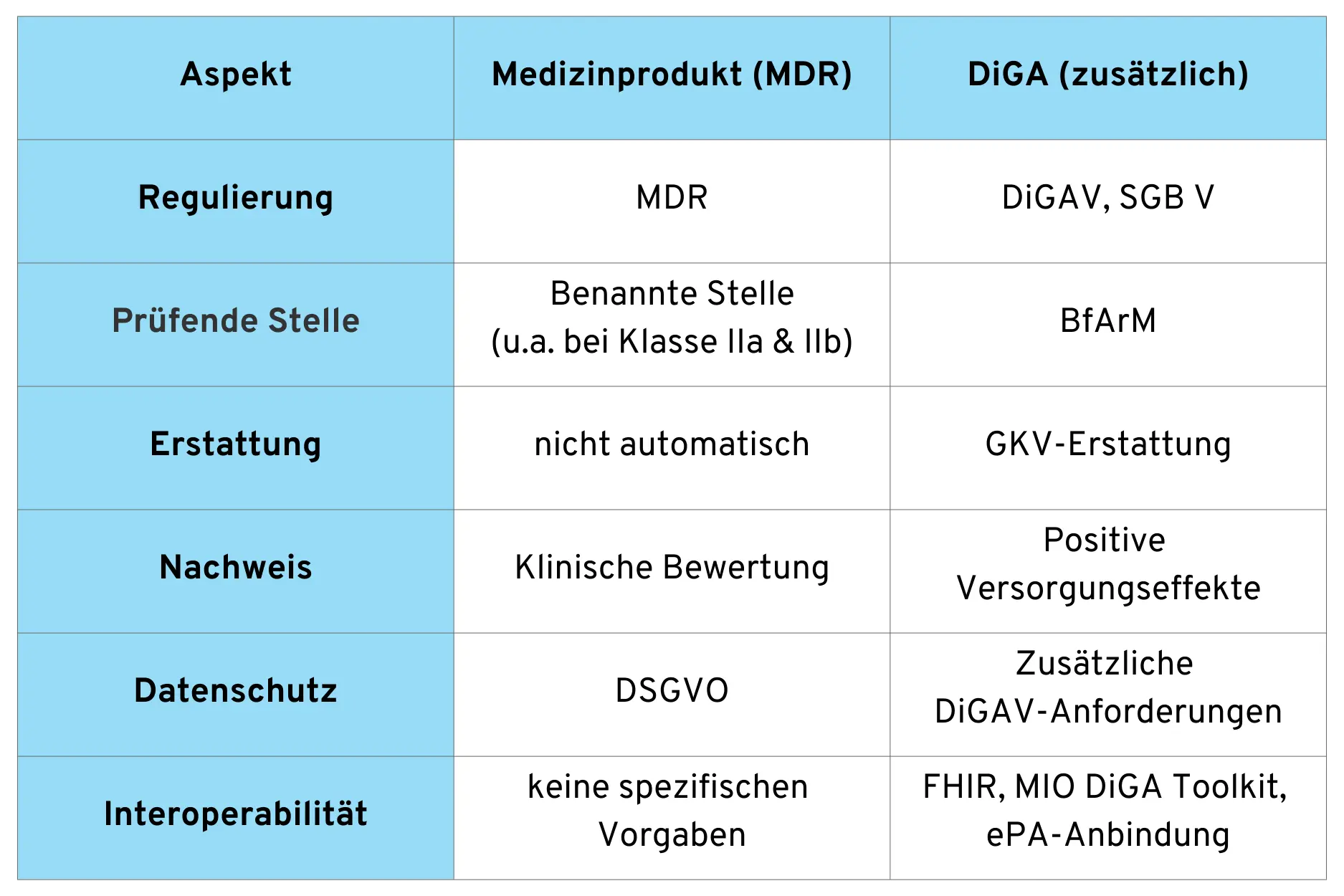
5. Unterschiede zwischen Medizinprodukt und DiGA
Nachdem wir die einzelnen Aspekte im Detail betrachtet haben, fassen wir einige wesentliche Unterschiede zwischen einem „normalen“ Medizinprodukt und einer DiGA zusammen.
 (Klicken zum Vergrößern)
(Klicken zum Vergrößern)
Diese Liste könnte noch sehr stark ausgeweitet werden und dient nur dem grundlegenden Verständnis.
6. Fazit: Empfehlungen für Hersteller
Der Weg zur DiGA führt immer über das Medizinprodukt. Wer eine digitale Gesundheitsanwendung entwickelt und in die GKV-Erstattung bringen möchte, muss beide regulatorischen Ebenen von Anfang an mitdenken.
Unsere Empfehlungen für Hersteller:
- Frühzeitig die regulatorische Strategie festlegen: Klären Sie zu Beginn, ob Ihr Produkt ein Medizinprodukt ist und ob der DiGA-Weg sinnvoll ist. Nicht jedes Medizinprodukt muss eine DiGA werden.
- MDR-Konformität als Basis sicherstellen: Ohne CE-Kennzeichnung keine DiGA. Planen Sie ausreichend Zeit für die Konformitätsbewertung ein. Das gilt insbesondere bei Klasse IIa und IIb, wo Benannte Stellen einbezogen werden müssen.
- DiGAV-Anforderungen parallel planen: Datenschutz, Datensicherheit und Interoperabilität sollten von Beginn an in die Produktentwicklung einfließen. Nachträgliche Anpassungen sind aufwendig und teuer.
- Studienplanung für den pVE-Nachweis frühzeitig starten: Insbesondere wenn Sie eine dauerhafte Aufnahme anstreben, beginnen Sie früh mit der Konzeption der Nachweisstudie.
- Beratung beim BfArM nutzen: Das BfArM bietet kostenpflichtige Beratungsgespräche an. Diese Investition lohnt sich, um Unsicherheiten im Vorfeld zu klären.
Sie planen eine DiGA? Wir entwickeln digitale Gesundheitsanwendungen im Auftrag für Unternehmen: von der Konzeption über die MDR-Zulassung bis zur Aufnahme ins DiGA-Verzeichnis. Wir übernehmen alle Schritte bis zur erfolgreichen Erstattung und danach.



