

Wie funktioniert die Medizinprodukt-Zulassung für Software? Wie lange dauert dieser Prozess? Und was sind die Kosten, die für die Zertifizierung anfallen? Der Weg zur Medizinprodukt-Zulassung ist auch für Software-Produkte komplex. Daher ist es ratsam, dass Sie sich früh genug mit der (Selbst-)Zertifizierung Ihrer Software beschäftigen und sich über die notwendigen Schritte klar werden. Schließlich gibt es auch abseits der reinen Software-Entwicklung einiges zu beachten. Wenn Sie erfahren möchten, was genau Sie für die Zulassung Ihrer Software als Medizinprodukt benötigen, um die CE-Kennzeichnung verwenden zu dürfen, sind Sie hier richtig. Der vorliegende Artikel thematisiert die Zulassung von Software-Medizinprodukten unter der Medical Device Regulation (MDR), welche die geltende Verordnung für Medizinprodukte in der EU ist.
Inhalt
- 1. Vorbereitung der Zertifizierung
- 2. Zulassung der Software
- 3. Rahmenbedingungen für die Zulassung von Software-Medizinprodukten
- 4. Fazit

Zeitplan für die Zulassung von Medizinprodukten
1. Vorbereitung der Zertifizierung
Die Vorbereitung auf die Zertifizierung beginnt in jedem Fall noch vor der eigentlichen Entwicklung Ihrer Medizinprodukt-Software. Denn der Aufwand, der für eine reibungslose Zulassung betrieben werden muss, ist stark von der Risikoklasse abhängig, welche Sie in den ersten Schritten des Projekts definieren. Diese entscheidet nämlich über das anzuwendende Konformitätsbewertungsverfahren und die Notwendigkeit einer benannten Stelle für die Zulassung. Was aber in allen Fällen notwendig ist, ist der Aufbau eines Qualitätsmanagementsystem. Dieses soll die Qualität Ihres Produkts sicherstellen und ist die Grundlage für die technische Dokumentation, welche für Medizinprodukte aller Risikoklassen erstellt werden muss.
1.1 Qualifizierung und Klassifizierung
Ganz zu Beginn einer jeden medizinischen Software steht die Definition des angestrebten Nutzens – genauer, die “Zweckbestimmung”. Denn diese entscheidet fundamental darüber, ob es sich bei dem geplanten Produkt überhaupt um ein Medizinprodukt handelt (Qualifizierung). Im darauffolgenden Schritt gilt es, das Produkt entsprechend zu klassifizieren und einer Risikoklasse (I, IIa, IIb oder III) zuzuordnen. Der vorliegende Artikel konzentriert sich auf die Zulassung und Zertifizierung Ihres Software-Medizinprodukts. Ein detaillierter Exkurs in die Qualifizierung und Klassifizierung von Software würde hier den Rahmen sprengen. Selbstverständlich haben wir zu diesen Themen eigene Artikel verfasst, welche Sie hier finden:
- Formulierung der Zweckbestimmung für (Software-)Medizinprodukte
- Leitfaden: Ist Ihre App ein Medizinprodukt?
- Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden
Wichtig ist, dass Sie die Klassifizierung Ihrer Software gewissenhaft durchführen und sich frühzeitig mit diesem Thema beschäftigen. Denn von der Risikoklasse hängt unter anderem das anzuwendende Konformitätsbewertungsverfahren ab, welches weiter unten in diesem Artikel thematisiert wird und den Aufwand, die Dauer und Kosten für das Projekt maßgeblich beeinflusst. Falls Sie noch kein Qualitätsmanagementsystem in Ihrem Unternehmen etabliert haben, gilt es im nächsten Schritt, ein solches aufzubauen (unabhängig von der Risikoklasse). Was Sie grundsätzlich bei der Medizinproduktentwicklung beachten müssen, erfahren Sie in diesem Artikel: Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten
1.2 Auswahl eines Konformitätsbewertungsverfahrens
„Konformitätsbewertung“ bezeichnet das Verfahren, nach dem festgestellt wird, ob die Anforderungen dieser Verordnung an ein Produkt erfüllt worden sind; (Zitat aus Artikel 2 MDR)
Damit Ihre Software eine CE-Kennzeichnung tragen darf, müssen Hersteller zuerst ein sogenanntes Konformitätsbewertungsverfahren durchführen. Die MDR bietet von Anhang IX bis XI drei verschiedene Bewertungsverfahren. Die Auswahl des anzuwendenden Verfahrens ist ebenfalls noch in der Planungsphase des Projekts zu treffen. An dieser Stelle sagen wir gleich, dass für Software eigentlich nur das Verfahren in Anhang IX praktikabel ist: Konformitätsbewertung auf der Grundlage eines Qualitätsmanagementsystems und einer Bewertung der technischen Dokumentation. Zwar ist es auch möglich, ein Bewertungsverfahren anhand einer sogenannten “Baumusterprüfung” durchzuführen, was aber in den meisten Fällen nicht sinnvoll ist. In einem Dokument (NB-MED/2.2/Rec4), das die Position der benannten Stellen zu diesem Thema angibt, steht wörtlich:
“[…] a pure product related evaluation without consideration of the design process is not considered adequate. Consequently, the use of some of the Conformity Assessment Procedures (CAP), as defined by the directives may be unsuitable for software.”
Ferner beschreibt das Dokument die Notwendigkeit von Lebenszyklusprozessen. Dies würde im Grunde bedeuten, dass die Produktentwicklung den Vorgaben der IEC 62304 folgen muss, welche ebenfalls ein Qualitätsmanagementsystem erfordert. Von daher stellt sich ohnehin die Frage, warum Hersteller dann nicht gleich das Konformitätsbewertungsverfahren nach Anhang IX wählen. Die Anwendung des Konformitätsbewertungsverfahrens in Anhang IX (und der Einbezug einer benannten Stelle) ist für Hersteller von Medizinprodukten der Klasse I nicht erforderlich. Das heißt aber nicht, dass Sie für ein Klasse I Produkt nichts machen müssen – ein Qualitätsmanagementsystem und die technische Dokumentation für Ihr Produkt benötigen Sie dennoch. Dies wird aber nicht von einer benannten Stelle überprüft. Doch welche Art von Software fällt überhaupt noch in Klasse I? Lesen Sie mehr dazu in diesem Artikel: Klassifizierung von Software-Medizinprodukten: MDR Leitfaden Neben dem Konformitätsbewertungsverfahren müssen Sie als Hersteller auch eine EU-Konformitätserklärung für Ihr Produkt unterzeichnen.
Info: Unterschied zwischen Qualitätsmanagmentsystem und technischer Dokumentation:
- Ein Qualitätsmanagementsystem beschreibt die Prozesslandschaft in Ihrem Unternehmen und ist produktunabhängig. Prozesse sind Arbeitsabläufe, die durch bestimmte Trigger angestoßen und durchlaufen werden und erzeugen immer einen oder mehrere Outcomes. Risikomanagement wäre ein klassisches Beispiel für einen Prozess (welcher natürlich auch in mehrere Teilprozesse untergliedert werden kann).
- Die technische Dokumentation ist hingegen produktspezifisch und sie enthält alle Dokumente und Aufzeichnungen, die das Produkt betreffen. Darunter fallen beispielsweise Pläne und Reports (z.B. für das Risikomanagement oder die klinische Bewertung).
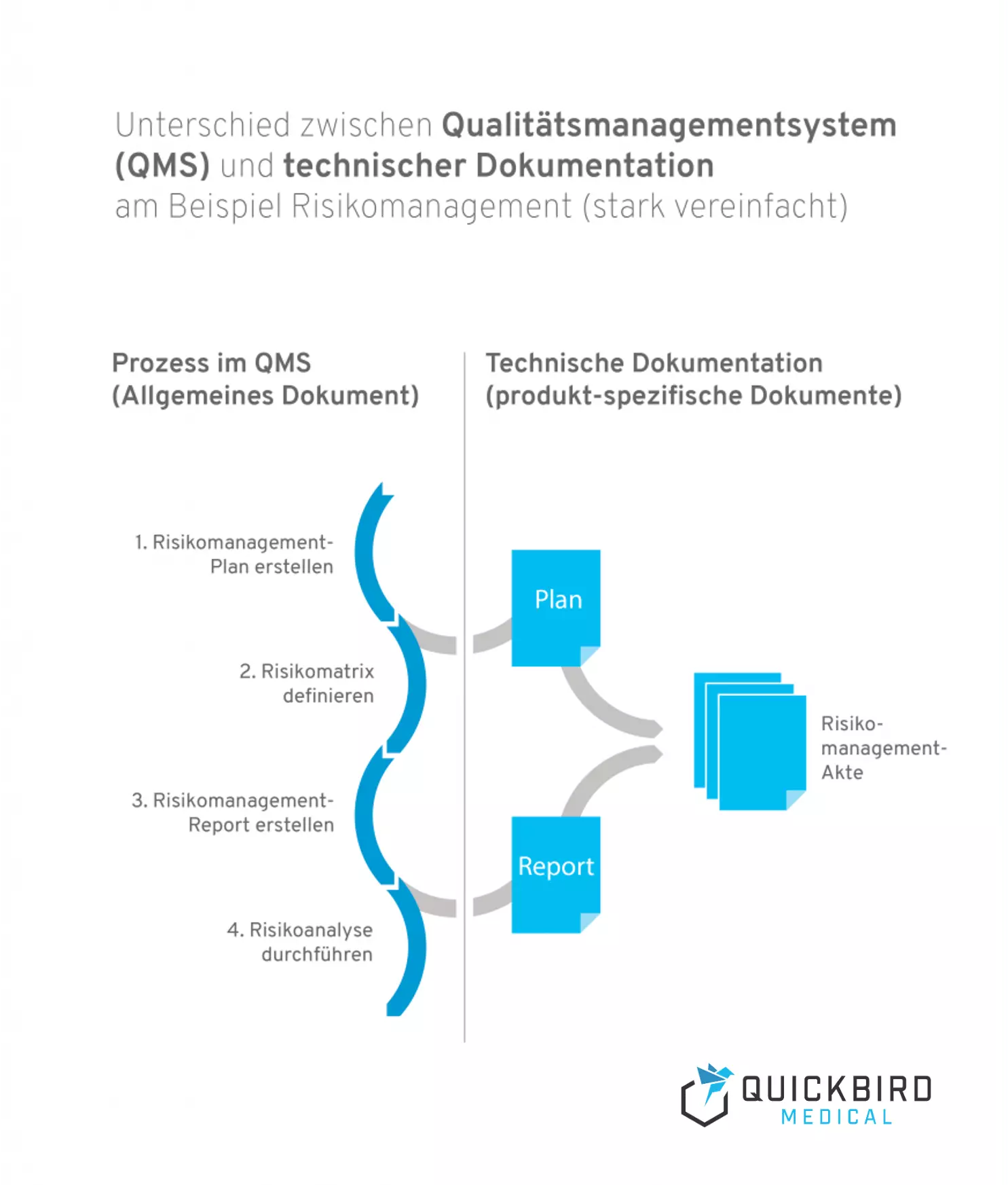
Unterschied zwischen Qualitätsmanagementsystem und technischer Dokumentation
1.3 Aufbau eines Qualitätsmanagementsystems
Bei Ihrem geplanten Produkt handelt es sich um ein Medizinprodukt? Glückwunsch, denn dann werden Sie bald auch ein eigenes Qualitätsmanagementsystem haben! Ein solches ist nämlich verpflichtend für alle Software-Medizinprodukthersteller und legt die Grundlage für den Großteil der technischen Dokumentation, die Sie gemäß MDR für Ihr Produkt erstellen müssen. Während die MDR keine wirklich gute Anleitung für den Aufbau eines QMS ist, halten Sie sich zunächst am besten an die ISO 13485 – das ist auch keine gute Anleitung, aber zumindest in Bezug auf die Anforderungen an vielen Stellen konkreter. Sie bekommen aber keinen Schritt-für-Schritt-Guide, der Ihnen sagt, was genau Sie machen müssen. Daher werden Sie sehr wahrscheinlich externe Unterstützung in Form von Beratungsdienstleistungen benötigen. Der Aufbau eines QMS nimmt je nach Unternehmensgröße und Komplexität bestehender Strukturen erfahrungsgemäß einige Monate in Anspruch. Achtung: Insgesamt sind die Anforderungen der MDR an Qualitätsmanagementsysteme etwas umfangreicher, als jene der ISO 13485. Die Norm legt aber eine gute Basis und deckt die meisten Anforderungen der MDR ab. Welche weiteren Normen Sie bei der Entwicklung von Software-Medizinprodukten berücksichtigen sollten, erfahren Sie in diesem Artikel: Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten.
1.4 Erstellung der technischen Dokumentation
Die technische Dokumentation ist in den Anhängen II und III der MDR spezifiziert. Sie soll Ihnen als Hersteller in erster Linie dabei helfen, bessere Produkte im Sinne der Leistungsfähigkeit und Patientensicherheit zu entwickeln und beinhaltet zum Beispiel Planungsdokumente, Berichte und Aufzeichnungen, die während des Produkt-Lebenszyklus entstehen. Zu berücksichtigen sind hier auch die Beschreibung geplanter Aktivitäten nach der Inverkehrbringung. Ein großer Teil der technischen Dokumentation geht aus den Prozessen Ihres Qualitätsmanagementsystems hervor, was bedeutet, dass vieles davon automatisch entsteht, wenn Sie sich an Ihre Prozesse halten. Die technische Dokumentation ist grob gesagt eine Sammlung aller Informationen, die das Produkt betreffen.
1.4.1 Durchführung einer klinischen Prüfung
In vielen Fällen müssen Sie für Ihr Produkt auch eine klinische Prüfung durchführen. Diese ist ebenfalls ein Teil der technischen Dokumentation und fällt in den Bereich der klinischen Bewertung. Mehr über die klinische Bewertung lesen Sie in diesem Artikel: MDR-Guide: Klinische Bewertung von Software-Medizinprodukten.
1.5 Suche nach einer benannten Stelle (für Klasse IIa oder höher)

Wann eine benannte Stelle notwendig ist
Info: Es kann auch seltene Ausnahmefälle geben, in denen auch für Medizinprodukte der Klasse I eine benannte Stelle erforderlich ist (z.B. wiederverwendbare chirurgische Instrumente). Das trifft auf Software aber in den allermeisten Fällen nicht zu. Produkte der Klasse IIa oder höher benötigen das “Go” einer benannten Stelle, bevor sie mit einer CE-Kennzeichnung auf den Markt gebracht werden können. Und spätestens hier kann die Planung der Medizinprodukt-Zertifizierung stark ins Schleudern geraten. Eine verspätete Suche nach einer benannten Stelle kann die Zulassung Ihrer Software nämlich um mehrere Monate verzögern – ein Szenario, das Sie als Medizinprodukt-Hersteller natürlich vermeiden möchten. Insgesamt gibt es 2 Hauptgründe, weshalb wir Ihnen empfehlen, sich frühzeitig auf die Suche zu begeben:
- Die Nachfrage ist größer als das Angebot: Wartezeiten von mehreren Monaten sind durchaus üblich. Konkret gibt es in Deutschland im Moment nur 7 benannte Stellen, die eine Prüfung nach MDR durchführen können (Stand März, 2022). Alle benannten Stellen nach MDR finden Sie im Nando-Verzeichnis.
- Nicht alle benannten Stellen haben eine gleich große Software-Affinität. Mit anderen Worten: manche Stellen machen Ihnen den Prozess unnötig schwer, da sie nicht verstehen, dass sich Software-Entwicklung von klassischer Medizinprodukt-Produktion in vielen Punkten unterscheidet. Es erspart Ihnen einiges an Zeit, und vor allem Nerven, wenn Sie einen Auditor erwischen, der mit Software-Produkten vertraut ist und “Ihre Sprache spricht”. Zudem verstehen Software-affine Stellen auch besser, welche Maßnahmen in einem QMS sinnvoll sind und die Chancen sind geringer, dass sie sich im Audit auf Prozesse fokussieren, die für Software wenig relevant sind.
Die Suche nach einer geeigneten benannten Stellen beginnt also in der Regel im oben verlinkten Nando-Verzeichnis. Wir möchten an dieser Stelle keine Empfehlung für oder gegen eine der benannten Stellen abgeben, aber folgende Quellen könnten bei der Auswahl einer benannten Stelle hilfreich sein:
- Wenn möglich, holen Sie sich Erfahrungsberichte von anderen Software-Medizinprdoukt-Herstellern ein
- Fragen Sie explizit nach Erfahrungen mit Software-Medizinprodukten in ersten Beratungsgesprächen mit den benannten Stellen
- Sie haben bereits ein ISO 13485 Zertifikat? Sehen Sie nach, ob Ihre Zertifizierungsstelle auch als benannte Stelle zertifiziert ist (vorausgesetzt, Sie sind mit dieser zufrieden)
- Sprechen Sie uns an
Falls Sie auch eine Zertifizierung nach ISO 13485 anstreben, macht es Sinn, diese bei einer Organisation zu beantragen, die auch als benannte Stelle zertifiziert ist – so bekommen Sie beides aus einer Hand, müssen Ihre Zertifizierungsstellen nicht dauernd wechseln und werden nicht Opfer von unterschiedlichen Interpretationen der einzelnen Organisationen.
2. Zulassung der Software
Sie haben ein Qualitätsmanagementsystem etabliert und Ihre technische Dokumentation erstellt? Super! In diesem Kapitel erfahren Sie, worauf Sie alles achten müssen, bevor Sie Ihr Produkt auf den Markt bringen können.
2.1 Prüfung durch die benannte Stelle (für Klasse IIa oder höher)
Wie schon weiter oben beschrieben, muss bei Produkten der Klasse IIa oder höher eine benannte Stelle für die Zulassung Ihres Medizinprodukts mit einbezogen werden. Dabei werden zwei Dinge (üblicherweise in dieser Reihenfolge) auditiert:
- Die technische Dokumentation
- Ihr Qualitätsmanagementsystem
Die Prüfung der technischen Dokumentation kann mehrere Feedbackschleifen beinhalten und sich je nach Bedarf auch über mehrere Wochen und Monate ziehen. Sobald die technische Dokumentation abgesegnet wurde, wird Ihr Qualitätsmanagementsystem auditiert (diese Reihenfolge ist üblich, aber nicht verpflichtend). Als Faustregel gilt: Je größer Ihr Unternehmen, desto länger dauert der Audit der benannten Stelle vor Ort. Stellen Sie sich also auf jeden Fall auf mehrere Tage ein, in denen die Auditoren zu Besuch kommen (in Ausnahmefällen wird der Audit auch Remote durchgeführt)).
Sie verfügen über ein ISO 13485 Zertifikat?
Rein formell macht eine ISO 13485-Zertifizierung keinen Unterschied – dadurch wird der Audit Ihres Qualitätsmanagementsystems durch die benannte Stelle weder verkürzt, noch fällt er ganz weg. Das liegt daran, dass der Scope der ISO 13485 nicht ganz deckungsgleich mit jenem der MDR ist, wenn es um Qualitätsmanagementsysteme geht. Zudem können Sie eine ISO Zertifizierung auch von Organisationen erhalten, die nicht als benannte Stelle zertifiziert sind. Dennoch ist ein ISO 13485 Zertifikat natürlich von Vorteil, weil Ihr Auditor mit einem ganz anderen Gefühl auf Sie zukommt, wenn ein solches vorhanden ist. Das signalisiert nämlich, dass Ihr Qualitätsmanagementsystem im Großen und Ganzen “schon in Ordnung” sein muss und die Wahrscheinlichkeit für grobe Abweichungen geringer ist. Zudem kommen Sie weniger in Erklärungsnot bei der Frage, warum sie glauben, dass Ihr Qualitätsmanagementsystem MDR-konform ist. Tipp: Falls Sie noch kein ISO 13485 Zertifikat für Ihr Qualitätsmanagementsystem haben, empfiehlt es sich, dieses im Zuge des QMS-Audits zu beantragen. Fragen Sie bei Ihrer benannten Stelle nach, ob das möglich ist. Das spart Ihnen möglicherweise einen zweiten Audit. Rechtlich gesehen benötigen Sie aber kein ISO 13485 Zertifikat.
2.1.1 Sonderfall bei Risikoklasse I
Wie bereits oben erwähnt, benötigen Hersteller von Klasse I Produkten keine benannte Stelle für die Konformitätsbewertung ihrer Software. Es reicht in diesem Fall, ein Qualitätsmanagementsystem zu etablieren, die technische Dokumentation nach Anhang II und III anzufertigen und im Anschluss die EU-Konformitätserklärung zu erstellen. Danach kann die Software als Medizinprodukt im DMIDS bzw. EUDAMED angemeldet werden (mehr dazu weiter unten) und Sie haben Ihre Pflichten als Hersteller erfüllt. Somit kann das Produkt guten Gewissens mit der CE-Kennzeichnung auf den Markt gebracht werden. Aber gibt es gar keine Prüfung von der Aufsichtsbehörde? Nicht zwangsläufig – zumindest ist der Markteintritt nicht davon abhängig. Offiziell sind Behörden nämlich nicht dazu verpflichtet, derartige Produkte direkt bei der Registrierung im Detail zu prüfen. Aber gerade bei der ersten Anmeldung eines Produkts ist es durchaus realistisch, dass die technische Dokumentation und/oder Ihr QMS zumindest stichprobenartig angeschaut wird. Stellen Sie sich also darauf ein, dass Sie einige Zeit nach der Anmeldung des Produkts von Ihrer Aufsichtsbehörde aufgefordert werden, ihr Einsicht in ihre technische Dokumentation zu gewähren (z.B. in das Risikomanagement oder die klinische Bewertung). Auf eine solche Anfrage müssen Sie aber nicht warten – Sie können Ihr Produkt bereits in Verkehr bringen. In diesem Artikel erfahren Sie, wie Sie die Risikoklasse Ihrer Medizinprodukt-Software bestimmen: Klassifizierung von Software-Medizinprodukten: MDR Leitfaden
2.2 Ihre EU-Konformitätserklärung
Vor dem Markteintritt erklären Sie als Hersteller die Konformität Ihres Produkts schriftlich. Dieses Dokument beinhaltet alle Informationen, die in Anhang IV der MDR definiert sind. Eine entsprechende Vorlage für die EU-Konformitätserklärung können Sie hier herunterladen.
EU-Konformitätserklärung – kostenloser Download
Wir senden Ihnen eine Vorlage für die EU-Konformitätserklärung kostenlos per E-Mail zu. Tragen Sie dazu einfach Ihre E-Mail-Adresse ein.
2.3 Registrierung des Medizinprodukts: EUDAMED und DMIDS
Muss ich mein Produkt beim BfArM (DMIDS) oder in der EUDAMED-Datenbank anmelden? Oder sogar in beiden Systemen? Diese Frage stellt sich im Moment allen Medizinprodukt-Herstellern, da geplant ist, dass EUDAMED das DMIDS in Zukunft ablöst. Im Moment ist sie recht schnell beantwortet: DMIDS ist Pflicht, EUDAMED freiwillig. Hinweis: Dieser Artikel beleuchtet den Stand vom 25.03.2022. Zwischenzeitliche Änderungen konnten nicht berücksichtigt werden.
3. Rahmenbedingungen für die Zulassung von Software-Medizinprodukten
3.1 Wie lange dauert die Zulassung?
Der Zeitaufwand für die Zertifizierung Ihres Medizinprodukts kann natürlich nicht pauschal bestimmt werden. Dennoch gibt es ein paar wichtige Parameter, die Sie in der Planung berücksichtigen müssen. Der in Klammern angegebene Zeitrahmen ist als Faustregel zu betrachten – in der Realität können diese Angaben auch schwanken:
- Aufbau eines Qualitätsmanagementsystems (Einige Monate)
- Produktentwicklung & Erstellung der technischen Dokumentation (Einige Monate bis Jahre)
- Klinische Prüfung, falls notwendig (Einige Monate bis Jahre)
- Für Klasse IIa oder höher:
- Suche nach einer benannten Stelle (bis zu 6 Monate)
- Prüfung der technischen Dokumentation und des QMS durch die benannte Stelle (ca. 3-12 Monate)Das heißt aber nicht, dass Sie für ein Klasse I Produkt nichts machen müssen – Sie müssen trotzdem ein QMS aufbauen und die technische Dokumentation erstellen. Dies wird aber nicht von einer benannten Stelle überprüft.
Bestimmt haben Sie gemerkt, dass die Notwendigkeit einer benannten Stelle der wichtigste Zeitfaktor ist. Das bedeutet, dass die Zulassung von Klasse I Medizinprodukten vergleichsweise kaum Zeit in Anspruch nimmt. Tipp: Wenn Sie eine benannte Stelle benötigen, können folgende Maßnahmen dazu beitragen, den Zeitaufwand möglichst gering zu halten:
- Kümmern Sie sich möglichst früh um einen Termin bei einer benannten Stelle zur Prüfung der technischen Dokumentation für Ihr Produkt. Natürlich kann nicht auf den Tag genau abgeschätzt werden, wann Ihre technische Dokumentation final ist. Aber es ist besser, einen geplanten Termin zu vereinbaren und diesen notfalls zu verschieben, als gar keinen zu haben. Vergessen Sie aber nicht, Ihre benannte Stelle unverzüglich darüber zu informieren, falls Sie den Termin nicht einhalten können.
- Entscheiden Sie sich für eine benannte Stelle, die eine gewisse Affinität für Software-Medizinprodukte aufweist. Das reduziert Kommunikationsschwierigkeiten und beugt unpraktischen Anpassungen Ihres QMS und anderer Dokumente vor.
- Achten Sie darauf, eine möglichst fehlerfreie technische Dokumentation einzureichen. Insbesondere Flüchtigkeitsfehler und offensichtliche Unstimmigkeiten sollten Sie vorab selbst beseitigen. Denn dadurch verkleinert sich die Zahl der Feedbackschleifen, welche in der Regel jeweils mehrere Tage oder Wochen dauern.
Insgesamt sollten Sie für die Prüfung der technischen Dokumentation und die Auditierung des Qualitätsmanagementsystems mehrere Monate einplanen.
3.2 Wie viel kostet die Zulassung?
Die Kosten für die Medizinprodukt-Zertifizierung können stark variieren, daher listen wir hier die Kostenpunkte auf, die üblicherweise am stärksten ins Gewicht fallen. Grundsätzlich lässt sich auch hier sagen: Mit einem Klasse I Produkt kommen sie am günstigsten weg. Denn ab Klasse IIa benötigen Sie eine benannte Stelle für die Konformitätsbewertung Ihrer Software, welche die Kosten natürlich erhöht. Nennenswert im Allgemeinen sind an dieser Stelle die Kosten für
- Beratungsleistungen beim Aufbau eines Qualitätsmanagementsystems
- UDI-Nummern für Produkte (üblicherweise wenige 100€/Jahr)
- Klinische Prüfung, falls notwendig (mehrere 10.000€ oder mehr)
- (nur für Klasse IIa oder höher:) die benannte Stelle (ca. 30.000 – 35.000€). Hier kommt es stark darauf an, wie “zufrieden” die benannte Stelle mit Ihrer technischen Dokumentation und dem Qualitätsmanagementsystem ist. Je mehr Aufwand (z.B. Feedbackschleifen) vonseiten der benannten Stelle notwendig ist, desto höher werden die Kosten. Auch die Unternehmensgröße und die Anzahl der Produkte sind ein entscheidender Faktor für die Kosten.
Wenn wir schon von Kosten sprechen, müssen wir neben den finanziellen Aufwänden natürlich auch den Faktor Zeit betrachten. Zum einen dauert der Aufbau eines Qualitätsmanagementsystems auf jeden Fall zumindest einige Monate. Dadurch entgehen Ihnen vermutlich auf kurze Sicht Umsätze – auch wenn Sie ein konformes Qualitätsmanagementsystem natürlich als sinnvolles Investment in die Zukunft sehen müssen. Schließlich ist ein solches das Fundament eines jeden Software-Medizinproduktherstellers. Ein weiterer Zeitfresser betrifft Hersteller von Produkten der Klasse IIa oder höher: eine benannte Stelle blockiert den Markteintritt von Medizinprdoukten immer um einige Monate. Das heißt, dass Sie nichts an dem Produkt verdienen können, bevor die Zertifizierung durch die benannte Stelle abgeschlossen ist. Überlegen Sie sich also vorab, ob diese Kosten für Ihr Unternehmen realistisch zu tragen sind. Insbesondere Startups unterschätzen den Aufwand oft und haben häufig nicht die notwendigen Ressourcen, um diese “umsatzfreie” Zeit zu überstehen.
4. Fazit
Als wichtigste Kernaussage lässt sich festhalten: Aufwand, Dauer und Kosten für die Zulassung Ihrer Medizinprodukt-Software stehen und fallen mit der Risikoklasse. Für alle Klassen gilt es aber, ein Qualitätsmangamentsystem aufzubauen und die technische Dokumentation für das Produkt anzufertigen. Der große Unterschied liegt darin, dass Sie als Hersteller ab der Risikoklasse IIa weitaus intensiver unter die Lupe genommen werden. Hersteller von Medizinprodukten der Klasse I werden mit ganz viel Glück erst viele Monate nach Markteintritt von ihrer Aufsichtsbehörde überprüft (und dann oft nur oberflächlich), wohingegen andere Hersteller sich von einer benannten Stelle zertifizieren lassen müssen. Diese überprüft sowohl die technische Dokumentation Ihres Produkts, als auch Ihr Qualitätsmanagementsystem. Bei den Kosten für die benannte Stelle sollten Sie mit etwa 30.000-35.000€ rechnen (je nach Aufwand können diese Kosten natürlich weitaus höher liegen) – vergessen Sie aber nicht die großen internen Aufwände, die für den Aufbau eines Qualitätsmanagementsystems betrieben werden müssen! Hier müssen Sie bestimmt einige Monate und viel Arbeitszeit investieren. Unterschätzen Sie diese Kosten nicht und seien Sie sich bewusst, dass es im Zuge der Medizinproduktzulassung auch zu unvorhergesehenen Verzögerungen von mehreren Monaten kommen kann, bevor Sie mit Ihrem Produkt erste Umsätze generieren. Folgende Artikel sind für Sie ebenfalls relevant, wenn Sie die Entwicklung eines Software-Medizinprodukts planen:
- Formulierung der Zweckbestimmung für (Software-)Medizinprodukte
- Leitfaden: Ist Ihre App ein Medizinprodukt?
- Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden
Noch mehr Artikel finden Sie in unserem Blog.

